
Abstract
Cannabis is among the most widely consumed psychoactive substances worldwide. Individual differences in cannabis use phenotypes can partly be explained by genetic differences. Technical and methodological advances have increased our understanding of the genetic aetiology of cannabis use. This narrative review discusses the genetic literature on cannabis use, covering twin, linkage, and candidate-gene studies, and the more recent genome-wide association studies (GWASs), as well as the interplay between genetic and environmental factors. Not only do we focus on the insights that these methods have provided on the genetic aetiology of cannabis use, but also on how they have helped to clarify the relationship between cannabis use and co-occurring traits, such as the use of other substances and mental health disorders. Twin studies have shown that cannabis use is moderately heritable, with higher heritability estimates for more severe phases of use. Linkage and candidate-gene studies have been largely unsuccessful, while GWASs so far only explain a small portion of the heritability. Dozens of genetic variants predictive of cannabis use have been identified, located in genes such as CADM2, FOXP2, and CHRNA2. Studies that applied multivariate methods (twin models, genetic correlation analysis, polygenic score analysis, genomic structural equation modelling, Mendelian randomisation) indicate that there is considerable genetic overlap between cannabis use and other traits (especially other substances and externalising disorders) and some evidence for causal relationships (most convincingly for schizophrenia). We end our review by discussing implications of these findings and suggestions for future work.
Similar content being viewed by others
Introduction
Cannabis is among the most widely consumed psychoactive substances worldwide. An estimated 4% of the world population aged 15 to 64 used cannabis at least once in 2019 [1]. While prevalences vary highly between countries, the overall European Union lifetime use prevalence is estimated to be 27.2% [2]. People mainly use cannabis to experience a psychoactive induced ‘high’ characterised by mild euphoria, relaxation, and perceptual and cognitive alterations [3]. These responses are likely related to the endogenous endocannabinoid system, given that Δ-9-tetrahydrocannabinol (THC) binds to cannabinoid receptors in different brain areas. Besides THC, an important component of cannabis is cannabidiol (CBD). By itself, CBD is not intoxicating (at typical doses) and has a much lower risk of adverse effects compared to THC [4]. This is confirmed by studies showing that cannabis with an elevated THC to CBD ratio is more damaging [5].
Indeed, a large body of research has demonstrated adverse effects linked to cannabis use. For example, cannabis use is associated with accidents, lower cognition and motivation, and suicide attempts and regular use has been related to various physical and psychological problems [5,6,7,8]. Regular use can also lead to addiction; in many countries cannabis is among the most common primary reasons for entering drug-related treatment [1] and cannabis use often precedes other drug use [9,10,11]. Problems related to cannabis use can in turn interfere with family, school, and work obligations [12]. Public health costs, law enforcement, and loss of work potential because of cannabis use are an economic drain on society [13]. In contrast, there may also be positive health benefits. There is some evidence that, by itself, CBD has antioxidant, anti-inflammatory, and neuroprotective properties [5]. Cannabinoid-based drugs are used to treat a range of medical conditions, including neurological disorders, psychiatric disorders, and pain [4, 14, 15]. While few serious side-effects have been reported, additional safety data are needed from more (and larger) clinical trials. In addition, it is important to note that non-medicinal CBD products (sold online or from health food retailers) lack quality standards and are not recommended for medicinal purposes [4].
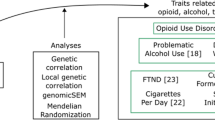
In light of the prevalence and adverse effects, for prevention, intervention and harm reduction efforts to be effective, it is important to understand why some individuals initiate cannabis use while others do not, and why a small subset progresses to regular user or develop a cannabis use disorder (CUD). In addition to environmental factors known to increase use (e.g. peer substance use, lower socio-economic status, poor neighbourhood characteristics, inadequate parental monitoring, high drug availability, and stressful life events [16,17,18,19,20]), risk of cannabis use runs also in families. A substantial part of the variability in cannabis use is due to genetic differences. This review provides an overview of current knowledge of the genetics of cannabis use, covering early twin studies to genome-wide association studies (GWASs) and post-GWAS analyses. When presenting results, we will refer to various indices of cannabis use, including initiation, frequency of use, and CUD which also be operationalised differently per study (Box 1 provides an overview of phenotypic definitions).
Twin studies
Before detailed information on the DNA sequence of the human genome was available, scientists were limited to studies using inferred genetic relatedness to explore the influence of genetic factors on cannabis use. Such studies relied on family, adoption, and twin designs. Family studies cannot distinguish between genetic and family environmental influences, and only few adoption studies were performed of cannabis use. A longitudinal adoption study showed that genetic influences on cannabis initiation were important at an early age (13–14 years old), but less so at age 17 and 18 [21].
Twin studies have proven more valuable because they typically used larger samples than adoption studies and they can differentiate between shared environmental and genetic influences. In twin studies, the resemblance between monozygotic twin pairs (who share all their DNA) is compared to that of dizygotic twin pairs (who share on average 50% of their segregated genes) [22]. If monozygotic twins resemble each other more than dizygotic twins on a certain trait, for example cannabis use, this is an indication that this trait is partly influenced by genetic difference between people. By applying sophisticated statistical models to twin data, it is possible to estimate what proportion of individual differences is due to genetic differences between people (heritability), shared (or family) environmental, and non-shared (or unique) environmental influences (see [23, 24]). Decades of twin studies have revealed that virtually every physical, behavioural, cognitive, and disease trait is heritable [25]. It is important to emphasise that heritability does not represent a fixed estimate nor does it describe individual levels of personal risk. Estimates of genetic and environmental variation are population estimates used to describe the sources of individual differences within a sample. When a trait or disorders is heritable, this does not mean that people’s outcomes are determined at conception; heritability does not equal genetic determinism. Instead, whether someone develops a certain disease or addiction depends on a complex interplay between genetic vulnerability and many environmental factors.
The heritability of various cannabis use phenotypes has been estimated in twin studies, most of which focussed on cannabis initiation or indices of CUD. A meta-analysis of these twin studies in 2010 [26] presented meta-analytic heritability estimates of 48% for females and 51% for males for cannabis initiation, and 51% for females and 59% for males for problematic cannabis use. In addition, Agrawal et al. [27] estimated a heritability of 35% and 27% for positive and negative subjective initial reactions to cannabis intake, Hines et al. [28] estimated that the opportunity to use cannabis was 64% heritable, and frequency of use 74%, and Minică et al. [29] estimated that age at first cannabis use was 38% heritable. In general, the relative genetic contribution is lower for the initiation of cannabis use compared to more severe stages of use such as problematic use, while for shared environmental influences, it was the other way around. Possibly, the initial stages of cannabis use are more sensitive to environmental factors, such as drug availability, peer influences, parental monitoring, and parental attitudes towards drug use, whereas the likelihood of progression to problematic use is more influenced by biological factors such as people’s physical response to THC intake. The pattern of higher heritability and lower family environmental influences for more severe phases of cannabis use has also been found for other substances [30, 31].
With multivariate twin methods [23], it is possible to estimate how much the genetic influences on one trait overlap with those underlying other traits. Multivariate twin studies have revealed that large portions of genetic factors in cannabis use initiation and problematic use are shared [32]. Correlations between measures of cannabis initiation, regular use, and problematic use suggest a single liability [33, 34], explained by common genes and environments [31, 32]. Similar patterns in terms of common genetic and environmental on different stages of use have also been observed for other substance [35, 36].
Multivariate twin studies have also explored to what extent genetic and environmental influences are shared across use of different substances. One study found that a common factor influenced by genetic factors, and family and non-family environmental influences underpins comorbid cannabis, sedative, stimulant, opioid, and psychedelic misuse [37]. Another study found that comorbid substance misuse (including cannabis, cocaine, hallucinogens, sedatives, stimulants, and opiates) is largely explained by overlapping genetic and shared environmental influences [38]. The same study also suggest that random environments determine how individuals choose to use a particular substance. A third study found that comorbid cannabis, cocaine, alcohol, caffeine, and nicotine misuse was best explained by two highly correlated genetic factors - one predisposing to cannabis and cocaine, the other to alcohol, caffeine, and nicotine misuse [39].
Overall, twin studies have demonstrated substantial overlap in genetic factors influencing earlier (experimental/regular use) and later (CUD) stages of cannabis use, and significant genetic overlap between use of cannabis and other substances. This general genetic vulnerability to substance use could be part of a much broader spectrum of personality characteristics or externalising psychopathology, characterised by substance use as well as conduct disorder, antisocial personality disorder, and other correlated traits [40,41,42,43,44,45].
Gene-finding studies
With the arrival of affordable DNA genotyping, the focus of behavioural genetics research shifted from family and twin studies to designs such as linkage analysis, candidate-gene studies, and GWASs, which rely on measured genotypes. Linkage analyses test for co-inheritance of genetic markers and traits within families. The segregation of a genetic marker within families is compared with the segregation of the trait in the family members. Downsides of this approach are that the analysis requires pedigree data and that linkage peaks only provide a rough indication of the implicated genomic region. In genome-wide linkage studies of cannabis use, most linkage peaks did not meet significance, and nearly all failed to replicate [46,47,48,49,50,51]. Ehlers et al. [48] found genome-wide significant linkage peaks for symptoms of cannabis dependence on chromosome 16 and 19, and in another study [49] on chromosomes 1, 3, 6, 7, and 9 for craving and cannabis symptoms. Hopfer et al. [50] reported suggestive evidence for linkage peaks for cannabis dependence symptoms on chromosome 3 and 9 in an adolescent sample. Han et al. [51] found a (non-significant) linkage peak at chromosome 8 for cannabis dependence; they then performed an association analysis under this peak, and found a significant and replicable association between variants in NRG1 and cannabis dependence. Non-significant peaks were reported on chromosome 14 for cannabis dependence symptoms [46], on chromosome 18 for cannabis frequency of use and initiation and chromosome 19 for early onset of cannabis use [47], and on chromosome 1 and 4 for cannabis problems [52]. The latter peak was in the region of the gamma-aminobutyric acid type A gene cluster, which includes GABRA2 that had previously been implicated in drug use disorders [53, 54].
Around the same time researchers also turned to candidate-gene studies, a hypothesis-driven method designed to tests for a correlation between a phenotype and a gene that is hypothesised to relate to this phenotype. For cannabis use these studies focused initially on variants in the cannabinoid receptor (CNR1) gene, located at chromosome 6. CNR1 is densely expressed in the central nervous system, notably in brain circuits thought to be important for reward and mnemonic processes related to substance misuse [55]. CNR1 was among the strongest candidate genes for cannabis use because it was known to be activated not only by endocannabinoids, but also plant phytocannabinoids such as THC, and synthetic analogs of THC. Using the candidate-gene approach, Hopfer et al. [56] and Agrawal et al. [57] found a significant association between the CNR1 gene and (symptoms of) cannabis dependence, but others could not replicate this association [58, 59]. In a meta-analysis, Benyamina et al. [60] showed a small but significant effect for the CNR1 AAT polymorphism on measures of substance dependence that included cannabis. Candidate-gene studies have also reported associations between cannabis use phenotypes and GABRA2 [53, 61], FAAH [62,63,64], and ABCB1 [65]. However, these associations largely failed to replicate [66,67,68]. For a comprehensive overview of candidate-gene studies for CUDs, see [69].
Overall, linkage and candidate-gene association studies were largely unsuccessful at identifying replicable genes. This failure is likely attributable to variation in research designs and phenotyping, lack of power, and publication bias [70]. Fortunately, technological advances permitted genome-wide analysis of genetic variants associated with complex traits, using GWASs. GWASs use genetic markers (typically single nucleotide polymorphisms (SNPs)) spanning the entire genome to systematically test for association with a trait. This approach has become a widely adopted method of identifying genetic associations.
The first cannabis GWASs, focussed on initiation [71, 72], dependence [73], and age at initiation [72], comprised small sample sizes and failed to identify genome-wide significant genetic loci. To increase power, large-scale collaborative efforts were undertaken. In 2012, the International Cannabis Consortium (ICC) was established with the aim of combining data from multiple cohorts to identify genetic variants associated with cannabis use. To date, the ICC has published three GWAS meta-analyses. The first [74] investigated cannabis initiation and involved a meta-analysis of 13 cohorts (N = 32,330, plus four replication samples (N = 5,627)). Although no individual SNP reached genome-wide significance, subsequent gene-based tests of association identified four genes significantly associated with cannabis initiation: NCAM1, CADM2, SCOC, and KCNT2. In a more recent ICC report [75], where the meta-analytic sample for cannabis initiation was increased to ~184,000 individuals, eight independent genome-wide significant SNPs in six regions were identified, as well as 35 significant genes in a gene-based tests of association. The third ICC GWAS report investigated age at onset of cannabis use (N = 24,953 individuals [29]), and identified a genome-wide significant association with SNPs in the ATP2C2 gene.
In 2016, Sherva et al. [76] identified the first genome-wide significant associations for cannabis dependence. The performed a GWAS for cannabis dependence criterion count in three substance dependence cohorts (N = 14,754 African American and European American participants; 18–36% cases). Three independent genome-wide significant SNPs were identified, two specific to African American participants (one in RP11-206M11.7 and one 12.4 kb upstream from the S100B gene) and one in the combined sample (in the CSMD1 gene). Two additional meta-analytic efforts for cannabis use disorder have been undertaken by (i) the Initiative for Integrative Psychiatric Research (iPsych) and deCODE genetics [77] and (ii) the Psychiatric Genetics Consortium—Substance Use Disorder (PGC-SUD) workgroup [78]. Demontis et al. [77] performed a GWAS for CUD with a discovery sample of 2,387 cases and almost 50,000 controls (plus a replication sample of 5,501 cases and ~300,000 controls). They identified one genome-wide significant risk locus for CUD, a SNP that is a strong marker for CHRNA2 expression. More recently, The PGC-SUD GWAS meta-analysis study based on 20,916 cases and 363,116 controls [78] identified two genome-wide significant loci: one novel locus in the FOXP2 gene, and the previously identified locus near CHRNA2 (and EPHX2). A systematic review of all cannabis use GWASs can be found elsewhere [79].
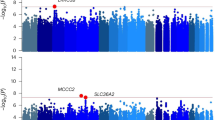
Whole genome sequencing (WGS) allows for more comprehensive association analysis than microarray-based GWASs, with the potential to identify rarer genetic variants. Gizer et al. [80] applied low-pass WGS to identify low frequency variants involved in cannabis dependence across two cohorts: a Native American tribal community and a family-based sample of primarily European ancestry. Their set-based analysis yielded two significant regions: a protein-coding region, C1orf110, and a regulatory region within the MEF2B gene. An overview of significant SNP and gene-based associations from the GWAS and WGS reports can be found in Table 1 and Fig. 1.

Illustration of the genomic locations of the genome-wide significant SNPs and genes for cannabis use phenotypes as identified in genome-wide association studies.
Revealing molecular mechanisms using functional annotation analyses
GWASs alone cannot inform the cascade of biological changes linking SNPs to cannabis use. This can, however, be addressed using gene-expression analyses via analysis of expression quantitative trait loci (eQTLs) or SNPs regulating gene-expression. Because gene-expression plays a critical role in human diseases [81], and because eQTLs regulate gene-expression, they likely provide a direct link between GWAS results and gene-expression studies [82]. Furthermore, eQTL analysis can discern transcriptome adaptations, while eQTLs in transcription factor binding sites, splice sites, and regulatory regions can reveal mechanisms by which genetic variants contribute to cannabis use [83]. Since most variants reside outside protein-coding regions, the influence of eQTLs on cell functioning likely involves subtle modification of gene transcription and translation [84]. By assessing eQTLs in linkage disequilibrium with SNPs associated with cannabis use, can we begin to explain their function.
Many of the genetic variants associated with cannabis use are located in non-protein-coding regions. Therefore, functional annotation analyses are required to elucidate downstream biological consequences underlying these genetic associations. Several methods have been developed for the biological interrogation of genetic associations [85]. The majority are based on the premise that associated SNPs influence disease risk by their influence on an intermediate molecular trait (known as a quantitative trait locus), such as gene expression, protein expression, exon splicing, or DNA methylation.
Functional annotation analyses of cannabis use are relatively sparse as only a handful studies have revealed significant genome-wide associations. However, the results reveal interesting leads to putative causal genes (Table 1). Multiple studies have explored if associated genetic variants regulate gene expression by browsing databases of expression Quantitative Trait Loci (eQTLs). Demontis et al. [77] found that genetic variants linked to CUD are eQTLs for CHRNA2, a nicotinic acetylcholine receptor gene. This finding was confirmed in the larger PGC-SUD GWAS meta-analysis [78] (including the Demontis sample). Given the association between CHRNA2 and cigarette smoking [86], Demontis et al. [77] explored whether the association between this gene and CUD was due to smoking as a confounding factor. Their results suggest that the signal is primarily driven by CUD.
Transcriptome-wide association study (TWAS) combines eQTL information across SNPs and tests the association between imputed levels of gene expression and disease risk to prioritise risk genes in a tissue-specific manner [87, 88]. Using the TWAS approach, Demontis et al. [77] found an association between CUD and CHRNA2 expression in the cerebellum, whereas Johnson et al. [78] found significant associations between CUD and expression levels for NAT6 (amygdala, cortex, frontal cortex), HYAL3 (multiple brain tissues), and IFRD2 (cerebellum). Significant associations were also reported between CUD and expression in NAT6, HYAL3, SHTN1, and FOXP2 in other tissues such as whole blood and adipose [78], highlighting the potential for non-invasive predictive bio-markers of CUD.
A TWAS of cannabis initiation [75] revealed 21 genes of which imputed expression levels are associated with initiation. The top association was found for CADM2; genetic variants associated with increased liability to initiate cannabis use are predicted to upregulate expression levels in eight non-brain tissues, including whole blood. CADM2 has been found to be associated with risk-taking, impulsivity, several measures of substance use, risky sexual behaviour, and self-control [89,90,91,92,93,94,95], suggesting that the association with cannabis use is part of a spectrum of externalising traits.
Agrawal et al. [96] conducted an extensive exploration of the molecular mechanisms underlying the association between rs1409568 and cannabis dependence. Based on its regulatory effects, this SNP was identified as the most plausible functional candidate within a locus at chromosome 10. The SNP appears to be located within an active enhancer and was predicted to bear active enhancer marks in several brain-derived tissues (e.g. dorsolateral prefrontal cortex). The risk increasing C allele is associated with reduced binding of several transcription factors. There was some support for this SNP to be associated with CpG methylation of TIAL1, with lower methylation scores in C allele carriers. Finally, the C allele of rs1409568 was also associated with a modest increase in right hippocampal volume (2.13%) in a sample of college students of whom very few met criteria for cannabis dependence. Of note, the counterintuitive finding of increased rather than decreased volumes was replicated in the phenotypic analysis.
Post-GWAS approaches
As with nearly all complex traits, GWAS has likewise revealed that cannabis use is a highly polygenic behaviour whereby individual differences are explained by many genetic variants each with very small effects. These tiny individual differences combined explain considerable amounts of genetic variation, but current GWASs capture only a fraction of the estimated heritability reported by twin studies. For instance, SNP-based heritability estimates are 11% for cannabis initiation [75], 3.6% for age at initiation [29], and between 6.7 and 12.1% (depending on the estimated population prevalence) for cannabis dependence [78]. The discrepancy in heritability reported by twin studies and GWASs is referred to as ‘missing heritability’, and is a known phenomenon in complex traits [97]. Among the various explanations proposed, missing heritability may arise from rare variants not captured by SNP arrays used in GWASs, or the poor ability of current genotyping arrays to capture structural variants. It is also possible that there may be interplay between genes and the environment not captured with the current GWAS design. As study sample sizes and genomic coverage increase, the expectation is for SNP-heritability to increase. Despite the small individual effect sizes and low SNP-heritabilities, summary-level data from GWASs—containing the association estimates of each genetic variant with the outcome variable—can be used for a range of useful secondary analyses. It is anticipated that this research will improve our understanding of the genetic architecture of cannabis use, and will help elucidate the nature of the relationships between cannabis use and comorbid complex traits including mental health outcomes.
Polygenic score analyses
Polygenic scores (PGSs) are predictors of the genetic liability of an individual to a disease or trait, and can be calculated by summing an individual’s ‘risk’ alleles for a certain phenotype weighted by the allele effect size, which are typically derived from effect estimates from large-scale GWASs. While PGSs only capture a small part of the genetic contribution to a trait, the validity of PGSs to predict complex psychiatric behaviours has been well demonstrated for many traits (e.g. [98,99,100]). Since the publications of the large-scale GWASs on cannabis initiation, age at initiation, and CUD, a number of studies have used the summary statistics to create PGSs in independent samples to predict observed cannabis use, other substance use, or correlated phenotypes. Several studies have found that cannabis PGSs significantly predict cannabis use phenotypes [77, 101,102,103,104,105] and mental health problems including depression and self-harm [103, 106], whereas other PGS analyses have not yielded significant results [105, 107, 108]. With larger samples we can determine if such discrepancies stem from lack of statistical power.
Using GWAS results to examine genetic correlations between traits
The introduction of affordable genotyping has meant that twin-based findings regarding sources of comorbidity of cannabis use with use of other substances or correlated traits can now be tested using measured genotypes. To assess shared genetic risks, linkage disequilibrium score regression (LDSR) can be used to compute genetic correlations between traits using summary-level GWAS data [109]. Such genetic correlation reflects the degree to which effects of genetic variants across the genome on one trait correlate with those on a second trait. Genetic correlations between cannabis use and various relevant other traits are shown in Fig. 2. Strong genetic correlations are found between cannabis use and other substance use. Cannabis initiation is strongly correlated with smoking initiation, whereas CUD is strongly correlated with dependency, e.g. alcohol dependence and cocaine dependence. This suggests a common genetic liability for initiating an addictive substance, and a partly distinct genetic liability for progressing from initiation to heavier use. Initiating substance use is most likely influenced by genetic factors that relate to externalising traits such as impulsivity. In line with this risk-taking and ADHD show some of the strongest genetic correlations with cannabis use. Cannabis use is also considerably correlated with major mental health disorders, e.g. major depressive disorder, schizophrenia, and bipolar disorder. Overall, these patterns imply that cannabis use has a considerable shared genetic aetiology with mental health problems. Note that while for cannabis initiation there are positive genetic correlations with intelligence, educational attainment, and income, these genetic correlations are negative for CUD.

The genetic correlations were computed with LD Score regression and the GWAS summary statistics of the GWASs on these traits (see Supplementary Table 1 for references and sample sizes).
By itself, a genetic correlation does not inform about the mechanisms underlying the association. Genomic structural equation modelling (SEM [110]) addresses this gap by providing insights into the nature of genetic associations. Genomic SEM is an extension of LDSR used to estimate genetic covariance between multiple traits using GWAS data. By constructing latent variables, different types of models can be built and (sub)models can be compared to test which has the superior fit. A number of studies have used genomic SEM to investigate relationships between cannabis use and other traits by modelling a latent genetic factor structure. One study included different substance use traits, and identified a unidimensional addiction risk factor, in which cannabis use (together with opioid use disorder) demonstrated the largest loadings [111]. Two other studies looked at mental health variables more broadly, and both found that cannabis dependence is part of a larger (externalising) factor comprised of, among others, alcohol dependence, nicotine dependence, and ADHD [112, 113].
Using GWAS results for causal inference
While a genetic correlation could arise due to a shared genetic liability between trait X and trait Y (‘horizontal pleiotropy’), this is not the only possible explanation. If there are causal relationships, such that X causes Y, or Y causes X, this would also lead to genetic correlations (‘vertical pleiotropy’) [114]. For example, if cannabis use causes schizophrenia, then genes underlying cannabis use should be indirectly associated with schizophrenia. Resolving the direction of causation may help improve preventive efforts. A genetic method that aims to infer causality using summary-level GWAS data is Mendelian randomisation (MR). To conduct an MR study, genetic variants that are strongly and reliably predictive of the proposed risk factor are typically required. Usually, this is achieved by selecting genetic variants that are genome-wide significantly (p < 5E-08) associated with the proposed risk factor in a well-powered GWAS. In some cases variants are selected based on a higher p-value threshold (e.g. p < 1E−07 or p < 1E−05). This is generally done when there is a lack of available genome-wide significant variants (note that this practice can lead to weak instrument bias). The selected variants are then employed as instrumental variables, or ‘proxies’, to test causal effects on an outcome. MR can be compared to a randomised clinical trial (RCT) in the sense that experimental randomisation into an ‘exposed’ and an ‘unexposed’ group is mimicked by the random assortment of a set of genetic variants. Genetic differences on these variants should not be (strongly) associated with confounders, which reduces bias [115].
There are important assumptions that need to be fulfilled to justify a causal interpretation of an MR analysis. The three main assumptions are that the genetic instrument must (1) be robustly associated with the exposure variable, (2), not be associated with any confounding variables, and (3) not influence the outcome through another path than through the exposure. Additional assumptions depending on the exact MR design are discussed elsewhere [116]. In general, it is preferable to use genetic instruments for which there is a (relatively) good understanding of how genetic variation leads to individual differences in the trait. For cannabis use, knowledge of biological pathways is limited and as mentioned before, there is evidence that the genetic variants involved are highly pleiotropic. This should be taken into account when judging evidence from MR studies looking at cannabis use. An important strength of MR is that a wide range of sophisticated sensitivity methods is available to assess the robustness of a causal finding.
MR studies have so far focused on two topics, the first being the relationship between cannabis use and the use of other substances. Three studies attempted to elucidate causal pathways of cannabis use with smoking, caffeine consumption, alcohol use, and other drug use (cocaine and opioid dependence), specifically trying to establish whether there is some kind of gateway mechanism. The first study (N = 38,181 to 112,117) found no clear evidence for causal relationships [117]. The second study (N = 25,153 to 207,726) found no evidence for causality except for one relationship: smoking initiation leading to higher caffeine intake [118]. The most recent MR study (N = 1749 to 1,232,091), which was also the most extensive with regards to the studied phenotypes and sensitivity methods, found evidence for causal effects of smoking initiation on cannabis initiation and cannabis dependence. In the other direction, they found evidence that cannabis initiation leads to smoking initiation, opioid dependence, and more alcohol consumption. The authors caution that these latter findings may indicate there is shared vulnerability rather than causality, because smoking and alcohol use typically begin before the use of the other substances (the temporality is unlikely) [119]. These findings emphasise that genetic variants for cannabis use, initiation specifically, are pleiotropic and likely not very specific in their effects.
The second focal point in MR literature is the relationship between cannabis use and mental health disorders. A recent systematic review paper summarised all MR studies that looked at substance use and mental health, including eight studies on cannabis use [120]. For major depression, self-harm behaviour, and cognitive functioning (N = 126,291 [121], 125,925 [106], and 3242 [122], respectively) there was no clear evidence for causal effects with cannabis initiation, in either direction. Note that the sample size of the analyses looking at cognitive functioning were underpowered. Between liability to schizophrenia and cannabis initiation there was evidence for bidirectional effects, based on three studies that used (partly) overlapping GWAS datasets (N = 79,845 [123], 32,330 to 150,064 [124], and 150,064 to 184,765 respectively [75]). Finally, based on two studies there was evidence that liability to ADHD increases the risk of cannabis initiation, without clear evidence for the reverse (N = 32,330 to 53,293 [125] and 53,293 to 184,765 [126]). Since this systematic review, other MR studies focussed on cannabis use have been published. One study found evidence that liability to bipolar disorder causally increases the risk of cannabis initiation, but no evidence for the reverse (N = 62,082 to 198,882; [127]). A second study found evidence that cannabis initiation causally increases the risk of suicide attempt (N = 50,264 to 162,082; [128]), while another found no evidence for causality between cannabis dependence and suicide-related behaviours (N = 18,223 to 117,733; [129]). Finally, a particularly comprehensive study investigated cannabis dependence and schizophrenia using multiple causally informative methods (genomic SEM, latent causal variable modelling, and MR) (N = 161,405 to 357,806; [130]). Some support was found for a causal influence of cannabis dependence on schizophrenia, but findings were not consistent across methods This last study is a nice demonstration of the importance of using several different methods to study (causal) relationships. This is referred to as ‘triangulation’, the premise being that if methods with different strengths and weaknesses point in the same direction, it is less likely a finding is an artefact [131]. Besides genetic methods, it is important for future studies to triangulate with alternative methods, such as longitudinal epidemiological analyses, or other types of (non-genetic) instrumental variable methods (e.g. population effects of cannabis policy changes).
Interplay between genetic vulnerability and environmental factors
Both genetic and environmental factors play a role in cannabis use. A complex interplay between these factors might determine individual differences in cannabis use and dependence. Interplay can occur as gene-environment interaction (G × E) where the effect of genetic vulnerability depends on the presence of environmental factors. For example, increased genetic risk for cannabis use may only influence patterns of use in people living in a neighbourhood where cannabis is widely available. Alternatively, genetic effects may reflect gene-environment correlations (rGE), where genetic liability to cannabis use influences environments to which individuals are either exposed or self-select into. For example, having an outgoing personality might lead to exposure to an environment where the use of cannabis is more common. Similarly, genetic effects could influence ones’ socio-economic status and thereby become correlated with one’s social surroundings and geographic location [132].
Rather than relying on candidate genes, G × E interaction studies now typically use polygenic measures [133]. A review of G × E studies using PGSs for substance use outcomes identified 34 publications (publication date before February 2018) but only three studies included cannabis outcome measures, and none used a cannabis use PGS. But since then, five studies have been published using a cannabis PGS to explore G × E interaction (Table 2). Two studies found significant PGS x Environment interactions; for trauma exposure [102] and for community activities [108]. Trauma seemed to exacerbate genetic risk for substance use, while engagement in community activities may serve as protective factor for cannabis use. Other environmental factors such as frequency of religious service attendance, organised sports, school activities, church activities and peer deviance were not or not consistently significant in these studies. The three other studies (exploring moderating roles for neighbourhood environment [134], peer cannabis use [101], prenatal stress, warm parenting, and cortisol reactivity [135]) did not find G × E interactions for cannabis use outcomes.
Regarding rGE, Johnson et al. [101] showed that individuals with high cannabis PGS are more likely to affiliate with cannabis using peers, a finding that is consistent with a process of social selection, whereby higher genetic risks for cannabis use may drive the propensity to affiliate with deviant drug using peers [136]. To our knowledge, only Pasman et al. (2019) have explicitly simultaneously modelled rGE (which was found to be absent) independently from G × E. Although G × E and rGE are typically studied independently, several statistical and conceptual reasons warrant joint assessment [137]. The presence of rGE may lead to false conclusions of G × E as many environmental factors are in fact influenced by genes themselves [137, 138].
In summary, evidence for G × E interactions for cannabis use is limited. Significant interaction need to be replicated and all studies used PGSs for cannabis initiation (based on [75]). Future studies should also evaluate G × E for more severe cannabis measures, but discovery GWAS samples for these phenotypes are still relatively small [79]. Furthermore, other environmental factors need to examined (for example parental factors) and the potential influence of rGE on G × E findings needs to be considered.
Clinical use
GWAS findings, the identification of mechanistic pathways, and studies investigating PGSs for cannabis use raise questions regarding the predictive validity of cannabis PGSs in clinical settings. Yanes et al. [139] have argued, broadly, that PGSs can be useful in terms of informing population screening programs, guiding therapeutic interventions, refining risk for individuals and families at high risk, and improving diagnosis. To date however, most cannabis research has been limited to basic science studies. While it is viable with PGSs to predict cannabis use in independent target samples, it is important to realise that PGSs currently contain too much noise and explain very little variation (up to a few percent), commensurate with other complex traits. Savatore et al. [140] have illustrated that although PGSs could be used to predict individuals and families meeting fewer clinical criteria for substance use disorders including cannabis, the effect sizes remain very small. Therefore, use of genetic results to identify individuals at risk of substance use disorders is modest at best, and future success depends upon increased and well phenotyped and genotyped samples [141]. It is not possible yet to use PGSs in clinical settings to meaningfully predict an individual’s genetic vulnerability to cannabis use. Efforts by the ICC and PGC-SUD workgroup to ascertain larger samples to improve the predictive validity of cannabis-based PGSs are ongoing. Furthermore, the modest heritability and importance of environmental risks shown by twin studies, suggests that clinical prediction algorithms will likely require a combination of measured genotypes and environments.
Future directions and conclusions
Insights into the genetic architecture of cannabis use are improving, but there are several steps we need to take in order to learn more [142]. Firstly, increasingly larger GWAS samples are required to capture more heritability. The genome coverage of GWASs also needs to improve to capture rare variants and other types of variation not captured by the current micro-arrays. Furthermore, we need to focus on including individuals of non-European ancestry. GWASs have been done almost exclusively in datasets of European ancestry. Systematic differences in ancestral genetic and environmental influences renders PGSs less useful in non-European samples. We need to improve the coverage of the population (e.g. non-European ancestry) to decrease the effect of ascertainment bias on the genetic signal. Environmental effects need to be accounted for in these genetic association studies by including within family and within region analyses while the interplay between genes and environment should be addressed more thoroughly. Twin studies show that genetic influences are more pronounced for cannabis dependence compared to initiation of cannabis use with similar SNP-based heritabilities from GWASs. Lastly, post-GWAS methodology needs to be further improved in order to disentangle the polygenic effects into underlying traits and underlying biological processes [142].
A specific point of focus in post-GWAS methodology is the improvement of MR and other causal inference methods. So far, the number of genetic variants associated with cannabis use—which are needed to use as instruments in an MR study—is limited. This may lead to weak instrument bias and spurious MR findings. In future studies, it is therefore recommended that evidence from a range of different MR methods is triangulated. Besides correcting for weak instrument bias [143], MR methods that allow correction for ‘correlated horizontal pleiotropy’ are important [144]. This phenomenon—whereby genetic variants affect two traits through a shared heritable factor—is highly relevant when testing relationships between cannabis use and mental health outcomes, but it is not taken into account in most common MR methods. Another promising approach is the MR direction of causation (MR-DoC) model, an adaptation which integrates the twin model with the MR design (a limitation is that well-powered twin samples are required) [145]. Besides genetic methods, it is important for future studies to also triangulate with alternative methods, such as longitudinal epidemiological analyses, or other types of (non-genetic) instrumental variable methods.
In conclusion, human genetics studies have provided a lot of insights of the genetic architecture of cannabis use. A large body of twin studies has shown that cannabis use is heritable—with moderate heritability for initiation of use and a somewhat higher heritability for measures of frequency and CUD. In the past decade, our insights into the molecular genetic architecture of cannabis use has also improved. Increases in sample size and technological advances have enabled GWASs to identify specific locations in the genome that are associated with cannabis use. So far, dozens of genetic variants and genes implicated in cannabis use have been reported, each explaining a tiny fraction of variance. Using summary-level GWAS data also provided us insight into the comorbidity between cannabis use and the use of other substance and mental health problems, providing evidence for shared genetic influences as well as some causal associations.
Future studies with increased sample sizes, including more diverse populations, higher genome coverage, and new approaches to improve the specificity of the genetic signals, should further increase our knowledge of the biological underpinnings of cannabis use and the predictive power of genetics.
References
United Nations Office on Drugs and Crime. World Drug Report 2021. 2021.
European Monitoring Centre for Drugs and Drug Addiction (EMCDDA). European Drug Report 2021: Trends and Developments. Luxembourg. 2021.
Green B, Kavanagh D, Young R. Being stoned: a review of self-reported cannabis effects. Drug Alcohol Rev. 2003;22:453–60.
Freeman TP, Hindocha C, Green SF, Bloomfield MAP. Medicinal use of cannabis based products and cannabinoids. BMJ 2019;365:l1141.
Ford TC, Hayley AC, Downey LA, Parrott AC. Cannabis: an overview of its adverse acute and chronic effects and its implications. Curr Drug Abus Rev. 2017;10:6–18.
Pearlson GD, Stevens MC, D’Souza DC. Cannabis and driving. Front Psychiatry 2021;12:689444.
Halladay JE, MacKillop J, Munn C, Jack SM, Georgiades K. Cannabis use as a risk factor for depression, anxiety, and suicidality: epidemiological associations and implications for nurses. J Addict Nurs. 2020;31:92–101.
Memedovich KA, Dowsett LE, Spackman E, Noseworthy T, Clement F. The adverse health effects and harms related to marijuana use: an overview review. CMAJ Open. 2018;6:E339–e46.
Fergusson DM, Horwood LJ. Does cannabis use encourage other forms of illicit drug use? Addiction 2000;95:505–20.
Lynskey MT, Vink JM, Boomsma DI. Early onset cannabis use and progression to other drug use in a sample of Dutch twins. Behav Genet. 2006;36:195–200.
Hall WD, Lynskey M. Is cannabis a gateway drug? Testing hypotheses about the relationship between cannabis use and the use of other illicit drugs. Drug Alcohol Rev. 2005;24:39–48.
Hasin DS. US epidemiology of cannabis use and associated problems. Neuropsychopharmacology 2018;43:195–212.
Hall W, Babor TF. Cannabis use and public health: assessing the burden. Addiction 2000;95:485–90.
Cohen K, Weizman A, Weinstein A. Positive and negative effects of cannabis and cannabinoids on health. Clin Pharmacol Therapeutics. 2019;105:1139–47.
Chesney E, Oliver D, Green A, Sovi S, Wilson J, Englund A, et al. Adverse effects of cannabidiol: a systematic review and meta-analysis of randomized clinical trials. Neuropsychopharmacology 2020;45:1799–806.
Prins SJ, Kajeepeta S, Hatzenbuehler ML, Branas CC, Metsch LR, Russell ST. School health predictors of the school-to-prison pipeline: substance use and developmental risk and resilience factors. J Adolesc Health. 2022;70:463–9.
Daniel JZ, Hickman M, Macleod J, Wiles N, Lingford-Hughes A, Farrell M, et al. Is socioeconomic status in early life associated with drug use? A systematic review of the evidence. Drug Alcohol Rev. 2009;28:142–53.
Myers B, McLaughlin KA, Wang S, Blanco C, Stein DJ. Associations between childhood adversity, adult stressful life events, and past-year drug use disorders in the National Epidemiological Study of Alcohol and Related Conditions (NESARC). Psychol Addictive Behav: J Soc Psychologists Addictive Behav. 2014;28:1117–26.
Olivares EL, Kendler KS, Neale MC, Gillespie NA. The genetic and environmental association between parental monitoring and risk of cannabis, stimulants, and cocaine initiation in a sample of male twins: does parenting matter? Twin Res Hum Genet. 2016;19:297–305.
Gillespie NA, Lubke GH, Gardner CO, Neale MC, Kendler KS. Two-part random effects growth modeling to identify risks associated with alcohol and cannabis initiation, initial average use and changes in drug consumption in a sample of adult, male twins. Drug Alcohol Depend. 2012;123:220–8.
Huibregtse BM, Corley RP, Wadsworth SJ, Vandever JM, DeFries JC, Stallings MC. A longitudinal adoption study of substance use behavior in adolescence. Twin Res Hum Genet. 2016;19:330–40.
Grasby KL, Verweij KJH, Mosing MA, Zietsch BP, Medland SE. Estimating heritability from twin studies. Methods Mol Biol. 2017;1666:171–94.
Neale MC, Cardon LR. Methodology for genetic studies of twins and families. Dordrecht: Kluwer; 1992.
Posthuma D, Beem AL, de Geus EJC, van Baal GCM, von Hjelmborg JB, Lachine I, et al. Theory and practice in quantitative genetics. Twin Res. 2003;6:361–76.
Polderman TJ, Benyamin B, de Leeuw CA, Sullivan PF, van Bochoven A, Visscher PM, et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet. 2015;47:702-9.
Verweij KJH, Zietsch BP, Lynskey MT, Medland SE, Neale MC, Martin NG, et al. Genetic and environmental influences on cannabis use initiation and problematic use: a meta-analysis of twin studies. Addiction 2010;105:417–30.
Agrawal A, Madden PA, Bucholz KK, Heath AC, Lynskey MT. Initial reactions to tobacco and cannabis smoking: a twin study. Addiction 2014;109:663–71.
Hines LA, Morley KI, Rijsdijk F, Strang J, Agrawal A, Nelson EC, et al. Overlap of heritable influences between cannabis use disorder, frequency of use and opportunity to use cannabis: trivariate twin modelling and implications for genetic design. Psychol Med. 2018;48:2786–93.
Minică CC, Verweij KJH, van der Most PJ, Mbarek H, Bernard M, van Eijk KR, et al. Genome-wide association meta-analysis of age at first cannabis use. Addiction 2018;113:2073–86.
Vink JM, Willemsen G, Boomsma DI. Heritability of smoking initiation and nicotine dependence. Behav Genet. 2005;35:397–406.
Agrawal A, Neale MC, Jacobson KC, Prescott CA, Kendler KS. Illicit drug use and abuse/dependence: modeling of two-stage variables using the CCC approach. Addictive Behav. 2005;30:1043–8.
Gillespie NA, Neale MC, Kendler KS. Pathways to cannabis abuse: a multi-stage model from cannabis availability, cannabis initiation and progression to abuse. Addiction 2009;104:430–8.
Gillespie NA, Neale MC, Legrand LN, Iacono WG, McGue M. Are the symptoms of cannabis use disorder best accounted for by dimensional, categorical, or factor mixture models? A comparison of male and female young adults. Psychol Addictive Behav: J Soc Psychologists Addictive Behav. 2012;26:68–77.
Kubarych TS, Kendler KS, Aggen SH, Estabrook R, Edwards AC, Clark SL, et al. Comparing factor, class, and mixture models of cannabis initiation and DSM cannabis use disorder criteria, including craving, in the Brisbane longitudinal twin study. Twin Res Hum Genet. 2014;17:89–98.
Fowler T, Lifford K, Shelton K, Rice F, Thapar A, Neale MC, et al. Exploring the relationship between genetic and environmental influences on initiation and progression of substance use. Addiction 2007;102:413–22.
Maes HH, Sullivan PF, Bulik CM, Neale MC, Prescott CA, Eaves LJ, et al. A twin study of genetic and environmental influences on tobacco initiation, regular tobacco use and nicotine dependence. Psychol Med. 2004;34:1251–61.
Tsuang MT, Lyons MJ, Meyer JM, Doyle T, Eisen SA, Goldberg J, et al. Co-occurrence of abuse of different drugs in men - The role of drug-specific and shared vulnerabilities. Arch Gen Psychiatry 1998;55:967–72.
Kendler KS, Jacobson KC, Prescott CA, Neale MC. Specificity of genetic and environmental risk factors for use and abuse/dependence of cannabis, cocaine, hallucinogens, sedatives, stimulants, and opiates in male twins. Am J Psychiatry. 2003;160:687–95.
Kendler KS, Myers J, Prescott CA. Specificity of genetic and environmental risk factors for symptoms of cannabis, cocaine, alcohol, caffeine, and nicotine dependence. Arch Gen Psychiatry. 2007;64:1313–20.
Krueger RF, Markon KE, Patrick CJ, Benning SD, Kramer MD. Linking antisocial behavior, substance use, and personality: an integrative quantitative model of the adult externalizing spectrum. J Abnorm Psychol. 2007;116:645–66.
Eaton NR, Krueger RF, Keyes KM, Skodol AE, Markon KE, Grant BF, et al. Borderline personality disorder co-morbidity: relationship to the internalizing-externalizing structure of common mental disorders. Psychol Med. 2011;41:1041–50.
Kendler KS, Prescott CA, Myers J, Neale MC. The structure of genetic and environmental risk factors for common psychiatric and substance use disorders in men and women. Arch Gen Psychiatry 2003;60:929–37.
Vink JM, Nawijn L, Boomsma DI, Willemsen G. Personality differences in monozygotic twins discordant for cannabis use. Addiction 2007;102:1942–6.
Agrawal A, Jacobson KC, Prescott CA, Kendler KS. A twin study of personality and illicit drug use and abuse/dependence. Twin Res. 2004;7:72–81.
Markon KE, Krueger RF, Watson D. Delineating the structure of normal and abnormal personality: an integrative hierarchical approach. J Personal Soc Psychol. 2005;88:139–57.
Agrawal A, Hinrichs AL, Dunn G, Bertelsen S, Dick DM, Saccone SF, et al. Linkage scan for quantitative traits identifies new regions of interest for substance dependence in the Collaborative Study on the Genetics of Alcoholism (COGA) sample. Drug Alcohol Depend. 2008;93:12–20.
Agrawal A, Morley KI, Hansell NK, Pergadia ML, Montgomery GW, Statham DJ, et al. Autosomal linkage analysis for cannabis use behaviors in Australian adults. Drug Alcohol Depend. 2008;98:185–90.
Ehlers CL, Gilder DA, Gizer IR, Wilhelmsen KC. Heritability and a genome-wide linkage analysis of a Type II/B cluster construct for cannabis dependence in an American Indian community. Addict Biol. 2009;14:338–48.
Ehlers CL, Gizer IR, Vieten C, Wilhelmsen KC. Linkage analyses of cannabis dependence, craving, and withdrawal in the San Francisco Family Study. Am J Med Genet B—Neuropsychiatr Genet. 2010;153B:802–11.
Hopfer CJ, Lessem JM, Hartman CA, Stallings MC, Cherny SS, Corley RP, et al. A genome-wide scan for loci influencing adolescent cannabis dependence symptoms: evidence for linkage on chromosomes 3 and 9. Drug Alcohol Depend. 2007;89:34–41.
Han S, Yang BZ, Kranzler HR, Oslin D, Anton R, Farrer LA, et al. Linkage analysis followed by association show NRG1 associated with cannabis dependence in African Americans. Biol Psychiatry 2012;72:637–44.
Agrawal A, Pergadia ML, Saccone SF, Lynskey MT, Wang JC, Martin NG, et al. An autosomal linkage scan for cannabis use disorders in the nicotine addiction genetics project. Arch Gen Psychiatry 2008;65:713–22.
Agrawal A, Edenberg HJ, Foroud T, Bierut LJ, Dunne G, Hinrichs AL, et al. Association of GABRA2 with drug dependence in the collaborative study of the genetics of alcoholism sample. Behav Genet. 2006;36:640–50.
Fehr C, Sander T, Tadic A, Lenzen KP, Anghelescu I, Klawe C, et al. Confirmation of association of the GABRA2 gene with alcohol dependence by subtype-specific analysis. Psychiatr Genet. 2006;16:9–17.
Zhang PW, Ishiguro H, Ohtsuki T, Hess J, Carillo F, Walther D, et al. Human cannabinoid receptor 1: 5 ‘ exons, candidate regulatory regions, polymorphisms, haplotypes and association with polysubstance abuse. Mol psychiatry 2004;9:916–31.
Hopfer CJ, Young SE, Purcell S, Crowley TJ, Stallings MC, Corley RP, et al. Cannabis receptor haplotype associated with fewer cannabis dependence symptoms in adolescents. Am J Med Genet B—Neuropsychiatr Genet. 2006;141B:895–901.
Agrawal A, Wetherill L, Dick DM, Xuei X, Hinrichs A, Hesselbrock V, et al. Evidence for association between polymorphisms in the cannabinoid receptor 1 (CNR1) gene and cannabis dependence. Am J Med Genet B—Neuropsychiatr Genet. 2009;150B:736–40.
Covault J, Gelernter J, Kranzler H. Association study of cannabinoid receptor gene (CNR1) alleles and drug dependence. Mol Psychiatry. 2001;6:501–2.
Heller D, Schneider U, Seifert J, Cimander KF, Stuhrmann M. The cannabinoid receptor gene (CNR1) is not affected in German i.v. drug users. Addict Biol. 2001;6:183–7.
Benyamina A, Kebir O, Blecha L, Reynaud M, Krebs MO. CNR1 gene polymorphisms in addictive disorders: a systematic review and a meta-analysis. Addict Biol. 2011;16:1–6.
Philibert RA, Gunter TD, Beach SR, Brody GH, Hollenbeck N, Andersen A, et al. Role of GABRA2 on risk for alcohol, nicotine, and cannabis dependence in the Iowa Adoption Studies. Psychiatr Genet. 2009;19:91–8.
Filbey FM, Schacht JP, Myers US, Chavez RS, Hutchison KE. Individual and additive effects of the CNR1 and FAAH genes on brain response to marijuana cues. Neuropsychopharmacology 2010;35:967–75.
Schacht JP, Selling RE, Hutchison KE. Intermediate cannabis dependence phenotypes and the FAAH C385A variant: an exploratory analysis. Psychopharmacology 2009;203:511–7.
Tyndale RF, Payne JI, Gerber AL, Sipe JC. The fatty acid amide hydrolase C385A (P129T) missense variant in cannabis users: Studies of drug use and dependence in Caucasians. Am J Med Genet B—Neuropsychiatr Genet. 2007;144B:660–6.
Benyamina A, Bonhomme-Faivre L, Picard V, Sabbagh A, Richard D, Blecha L, et al. Association between ABCB1 C3435T polymorphism and increased risk of cannabis dependence. Prog Neuro-Psychopharmacol Biol Psychiatry 2009;33:1270–4.
Haughey HM, Marshall E, Schacht JP, Louis A, Hutchison KE. Marijuana withdrawal and craving: influence of the cannabinoid receptor 1 (CNR1) and fatty acid amide hydrolase (FAAH) genes. Addiction 2008;103:1678–86.
Lind PA, Macgregor S, Agrawal A, Montgomery GW, Heath AC, Martin NG, et al. The role of GABRA2 in alcohol dependence, smoking, and illicit drug use in an Australian population sample. Alcohol-Clin Exp Res. 2008;32:1721–31.
Verweij KJH, Zietsch BP, Liu JZ, Medland SE, Lynskey MT, Madden PAF, et al. No association of candidate genes with cannabis use in a large sample of Australian twin families. Addict Biol. 2012;17:687–90.
Agrawal A, Lynskey MT. Candidate genes for cannabis use disorders: findings, challenges and directions. Addiction 2009;104:518–32.
Munafo MR. Reliability and replicability of genetic association studies. Addiction 2009;104:1439–40.
Verweij KJH, Vinkhuyzen AAE, Benyamin B, Lynskey MT, Quaye L, Agrawal A, et al. The genetic aetiology of cannabis use initiation: a meta-analysis of genome-wide association studies and a SNP-based heritability estimation. Addict Biol. 2013;18:846–50.
Minica CC, Dolan CV, Hottenga JJ, Pool R, Fedko IO, Mbarek H, et al. Heritability, SNP- and gene-based analyses of cannabis use initiation and age at onset. Behav Genet. 2015;45:503–13.
Agrawal A, Lynskey MT, Hinrichs A, Grucza R, Saccone SF, Krueger R, et al. A genome-wide association study of DSM-IV cannabis dependence. Addict Biol. 2011;16:514–8.
Stringer S, Minica CC, Verweij KJ, Mbarek H, Bernard M, Derringer J, et al. Genome-wide association study of lifetime cannabis use based on a large meta-analytic sample of 32 330 subjects from the International Cannabis Consortium. Transl psychiatry 2016;6:e769.
Pasman JA, Verweij KJH, Gerring Z, Stringer S, Sanchez-Roige S, Treur JL, et al. GWAS of lifetime cannabis use reveals new risk loci, genetic overlap with psychiatric traits, and a causal influence of schizophrenia. Nat Neurosci. 2018;21:1161–70.
Sherva R, Wang Q, Kranzler H, Zhao H, Koesterer R, Herman A, et al. Genome-wide association study of cannabis dependence severity, novel risk variants, and shared genetic risks. JAMA Psychiatry 2016;73:472–80.
Demontis D, Rajagopal VM, Als TD, Grove J, Pallesen J, Hjorthoj C, et al. Genome-wide association study implicates CHRNA2 in cannabis use disorder. Nat Neurosci. 2019;22:1066–74.
Johnson EC, Demontis D, Thorgeirsson TE, Walters RK, Polimanti R, Hatoum AS, et al. A large-scale genome-wide association study meta-analysis of cannabis use disorder. Lancet Psychiatry 2020;7:1032–45.
Hillmer A, Chawar C, Sanger S, D’Elia A, Butt M, Kapoor R, et al. Genetic basis of cannabis use: a systematic review. BMC Med Genomics. 2021;14:203.
Gizer IR, Bizon C, Gilder DA, Ehlers CL, Wilhelmsen KC. Whole genome sequence study of cannabis dependence in two independent cohorts. Addict Biol. 2018;23:461–73.
Schadt EE, Lamb J, Yang X, Zhu J, Edwards S, Guhathakurta D, et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet. 2005;37:710–7.
Franke L, Jansen RC. eQTL analysis in humans. Methods Mol Biol. 2009;573:311–28.
Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–7.
Shastry BS. SNPs: impact on gene function and phenotype. Methods Mol Biol. 2009;578:3–22.
Uffelmann E, Posthuma D. Emerging methods and resources for biological interrogation of neuropsychiatric polygenic signal. Biol Psychiatry 2021;89:41–53.
Liu M, Jiang Y, Wedow R, Li Y, Brazel DM, Chen F, et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019;51:237–44.
Gamazon ER, Wheeler HE. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47:1091–8.
Gusev A, Ko A. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48:245–52.
Morris J, Bailey MES, Baldassarre D, Cullen B, de Faire U, Ferguson A, et al. Genetic variation in CADM2 as a link between psychological traits and obesity. Sci Rep. 2019;9:7339.
Arends RM, Pasman JA, Verweij KJH, Derks EM, Gordon SD, Hickie I, et al. Associations between the CADM2 gene, substance use, risky sexual behavior, and self-control: a phenome-wide association study. Addict Biol. 2021;26:e13015.
Boutwell B, Hinds D, Tielbeek J, Ong KK, Day FR, Perry JRB. Replication and characterization of CADM2 and MSRA genes on human behavior. Heliyon 2017;3:e00349.
Sanchez-Roige S, Fontanillas P. Genome-wide association studies of impulsive personality traits (BIS-11 and UPPS-P) and drug experimentation in up to 22,861 adult research participants identify loci in the CACNA1I and CADM2. Genes 2019;39:2562–72.
Karlsson Linner R, Biroli P. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat Genet. 2019;51:245–57.
Karlsson Linnér R, Mallard TT, Barr PB, Sanchez-Roige S, Madole JW, Driver MN, et al. Multivariate analysis of 1.5 million people identifies genetic associations with traits related to self-regulation and addiction. Nat Neurosci. 2021;24:1367–76.
Pasman JA, Chen Z, Vink JM, Van den Oever MC, Pattij T, de Vries TJ, et al. The CADM2 gene and behavior: a phenome-wide scan in UK-Biobank. Behav Genet. 2022;52:306–14.
Agrawal A, Chou YL, Carey CE, Baranger DAA, Zhang B, Sherva R, et al. Genome-wide association study identifies a novel locus for cannabis dependence. Mol Psychiatry. 2018;23:1293–302.
Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet. 2011;88:294–305.
Murray GK, Lin T, Austin J, McGrath JJ, Hickie IB, Wray NR. Could polygenic risk scores be useful in psychiatry?: a review. JAMA Psychiatry. 2021;78:210–9.
Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009;460:748–52.
Andersen AM, Pietrzak RH, Kranzler HR, Ma L, Zhou H, Liu X, et al. Polygenic scores for major depressive disorder and risk of alcohol dependence. JAMA Psychiatry. 2017;74:1153–60.
Johnson EC, Tillman R, Aliev F, Meyers JL, Salvatore JE. Exploring the relationship between polygenic risk for cannabis use, peer cannabis use and the longitudinal course of cannabis involvement. Addiction. 2019;114:687–97.
Meyers JL, Salvatore JE, Aliev F, Johnson EC, McCutcheon VV, Su J, et al. Psychosocial moderation of polygenic risk for cannabis involvement: the role of trauma exposure and frequency of religious service attendance. Transl Psychiatry. 2019;9:269.
Hodgson K, Coleman JRI, Hagenaars SP, Purves KL, Glanville K, Choi SW, et al. Cannabis use, depression and self-harm: phenotypic and genetic relationships. Addiction 2020;115:482–92.
Brick LA, Benca-Bachman CE, Bertin L, Martin KP, Risner V, Palmer RHC. The intermediary role of adolescent temperamental and behavioral traits on the prospective associations between polygenic risk and cannabis use among young adults of European Ancestry. Addiction 2021;116:2779–89.
Paul SE, Hatoum AS, Barch DM, Thompson WK, Agrawal A, Bogdan R, et al. Associations between cognition and polygenic liability to substance involvement in middle childhood: results from the ABCD study. Drug Alcohol Depend. 2022;232:109277.
Lim KX, Rijsdijk F, Hagenaars SP, Socrates A, Choi SW, Coleman JR, et al. Studying individual risk factors for self-harm in the UK Biobank: a polygenic scoring and Mendelian randomisation study. PLoS Med. 2020;17:e1003137.
Allegrini AG, Verweij KJH, Abdellaoui A, Treur JL, Hottenga JJ, Willemsen G, et al. Genetic vulnerability for smoking and cannabis use: associations with E-cigarette and water pipe use. Nicotine Tob Res. 2019;21:723–30.
Thomas NS, Salvatore JE, Gillespie NA, Aliev F, Ksinan AJ, Dick DM. Cannabis use in college: genetic predispositions, peers, and activity participation. Drug Alcohol Depend. 2021;219:108489.
Bulik-Sullivan BK, Finucane HK, Anttila V, Gusev A, Day FR. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–41.
Grotzinger AD, Rhemtulla M, de Vlaming R, Ritchie SJ, Mallard TT, Hill WD, et al. Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat Hum Behav. 2019;3:513–25.
Hatoum AS, Johnson EC, Colbert SMC, Polimanti R, Zhou H, Walters RK, et al. The addiction risk factor: a unitary genetic vulnerability characterizes substance use disorders and their associations with common correlates. Neuropsychopharmacology 2021;47:1739–45.
Abdellaoui A, Smit DJA, van den Brink W, Denys D, Verweij KJH. Genomic relationships across psychiatric disorders including substance use disorders. Drug Alcohol Depend. 2021;220:108535.
Waldman ID, Poore HE, Luningham JM, Yang J. Testing structural models of psychopathology at the genomic level. World Psychiatry 2020;19:350–9.
van Rheenen W, Peyrot WJ, Schork AJ, Lee SH, Wray NR. Genetic correlations of polygenic disease traits: from theory to practice. Nat Rev Genet. 2019;20:567–81.
Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ (Clin Res ed) 2018;362:k601.
de Leeuw C, Savage J, Bucur IG, Heskes T, Posthuma D. Understanding the assumptions underlying Mendelian randomization. Eur J Hum Genet. 2022;30:653–60.
Verweij KJH, Treur JL, Vink JM. Investigating causal associations between use of nicotine, alcohol, caffeine and cannabis: a two-sample bidirectional Mendelian randomization study. Addiction 2018;113:1333–8.
Chang LH, Ong JS, An J, Verweij KJH, Vink JM, Pasman J, et al. Investigating the genetic and causal relationship between initiation or use of alcohol, caffeine, cannabis and nicotine. Drug Alcohol Depend. 2020;210:107966.
Reed ZE, Wootton RE, Munafò MR. Using Mendelian randomization to explore the gateway hypothesis: possible causal effects of smoking initiation and alcohol consumption on substance use outcomes. Addiction 2022;117:741–50.
Treur JL, Munafò MR, Logtenberg E, Wiers RW, Verweij KJH. Using Mendelian randomization analysis to better understand the relationship between mental health and substance use: a systematic review. Psychol Med. 2021;51:1593–624.
Howard DM, Adams MJ, Clarke T-K, Hafferty JD, Gibson J, Shirali M, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019;22:343–52.
Mahedy L, Wootton R, Suddell S, Skirrow C, Field M, Heron J, et al. Testing the association between tobacco and cannabis use and cognitive functioning: Findings from an observational and Mendelian randomization study. Drug Alcohol Depend. 2021;221:108591.
Vaucher J, Keating BJ, Lasserre AM, Gan W, Lyall DM, Ward J, et al. Cannabis use and risk of schizophrenia: a Mendelian randomization study. Mol Psychiatry 2017;23:1287–92.
Gage SH, Jones HJ, Burgess S, Bowden J, Davey Smith G, Zammit S, et al. Assessing causality in associations between cannabis use and schizophrenia risk: a two-sample Mendelian randomization study. Psychol Med. 2017;47:971–80.
Soler Artigas M, Sánchez-Mora C, Rovira P, Richarte V, Garcia-Martínez I, Pagerols M, et al. Attention-deficit/hyperactivity disorder and lifetime cannabis use: genetic overlap and causality. Mol Psychiatry 2020;25:2493–503.
Treur JL, Demontis D, Smith GD, Sallis H, Richardson TG, Wiers RW, et al. Investigating causality between liability to ADHD and substance use, and liability to substance use and ADHD risk, using Mendelian randomization. Addict Biol. 2021;26:e12849.
Jefsen OH, Speed M, Speed D, Østergaard SD. Bipolar disorder and cannabis use: a bidirectional two-sample Mendelian randomization study. Addict Biol. 2021;26:e13030.
Orri M, Séguin JR, Castellanos-Ryan N, Tremblay RE, Côté SM, Turecki G, et al. A genetically informed study on the association of cannabis, alcohol, and tobacco smoking with suicide attempt. Mol Psychiatry 2021;26:5061–70.
Colbert SMC, Hatoum AS, Shabalin A, Li QS, Coon H, Nelson EC, et al. Exploring the genetic overlap of suicide-related behaviors and substance use disorders. Am J Med Genet B—Neuropsychiatr Genet. 2021;186:445–55.
Johnson EC, Hatoum AS, Deak JD, Polimanti R, Murray RM, Edenberg HJ, et al. The relationship between cannabis and schizophrenia: a genetically informed perspective. Addiction 2021;116:3227–34.
Lawlor DA, Tilling K, Davey Smith G. Triangulation in aetiological epidemiology. Int J Epidemiol. 2017;45:1866–86.
Strom NI, Yu D, Gerring ZF, Halvorsen MW, Abdellaoui A, Rodriguez-Fontenla C, et al. Genome-wide association study identifies new locus associated with OCD. medRxiv. 2021. https://doi.org/10.1101/2021.10.13.21261078.
Vink JM. Genetics of addiction: future focus on Gene × Environment Interaction? J Stud Alcohol Drugs. 2016;77:684–7.
Pasman JA, Verweij KJH, Abdellaoui A, Hottenga JJ, Fedko IO, Willemsen G, et al. Substance use: Interplay between polygenic risk and neighborhood environment. Drug Alcohol Depend. 2020;209:107948.
Marceau K, Brick LA, Pasman JA, Knopik VS, Reijneveld SA. Interactions between genetic, prenatal, cortisol, and parenting influences on adolescent substance use and frequency: a TRAILS Study. Eur Addict Res. 2021;28:176–85.
Gillespie NA, Neale MC, Jacobson K, Kendler KS. Modeling the genetic and environmental association between peer group deviance and cannabis use in male twins. Addiction 2009;104:420–9.
Lau JY, Eley TC. Disentangling gene-environment correlations and interactions on adolescent depressive symptoms. J Child Psychol Psychiatry Allied Discip. 2008;49:142–50.
Pasman JA, Verweij KJH, Vink JM. Systematic review of polygenic gene-environment interaction in tobacco, alcohol, and cannabis use. Behav Genet. 2019;49:349–65.
Yanes T, McInerney-Leo AM, Law MH, Cummings S. The emerging field of polygenic risk scores and perspective for use in clinical care. Hum Mol Genet. 2020;29:R165–r76.
Salvatore JE, Barr PB. Sibling comparisons elucidate the associations between educational attainment polygenic scores and alcohol, nicotine and cannabis. Addiction. 2020;115:337–46.
Barr PB, Ksinan A, Su J, Johnson EC, Meyers JL, Wetherill L, et al. Using polygenic scores for identifying individuals at increased risk of substance use disorders in clinical and population samples. Transl Psychiatry 2020;10:196.
Abdellaoui A, Verweij KJH. Dissecting polygenic signals from genome-wide association studies on human behaviour. Nat Hum Behav. 2021;5:686–94.
Zhao Q, Wang J, Hemani G, Bowden J, Small DS. Statistical inference in two-sample summary-data Mendelian randomization using robust adjusted profile score. Ann Stat. 2020;48:1742–69. 28
Morrison J, Knoblauch N, Marcus JH, Stephens M, He X. Mendelian randomization accounting for correlated and uncorrelated pleiotropic effects using genome-wide summary statistics. Nat Genet. 2020;52:740–7.
Minică CC, Dolan CV, Boomsma DI, de Geus E, Neale MC. Extending causality tests with genetic instruments: an integration of mendelian randomization with the classical twin design. Behav Genet. 2018;48:337–49.
Yurko R, Roeder K, Devlin B, G’Sell M. H-MAGMA, inheriting a shaky statistical foundation, yields excess false positives. Ann Hum Genet. 2021;85:97–100.
Acknowledgements
K.J.H.V., A.A., and J.L.T. are supported by the Foundation Volksbond Rotterdam. J.L.T. is supported by a Veni grant from the Netherlands Organization for Scientific Research (NWO; grant number 016.Veni.195.016) and by a L’Oréal-UNESCO For Women in Science Award. A.A. is supported by grant 849200011 from The Netherlands Organisation for Health Research and Development. We would like to thank the research participants and employees of 23andMe, Inc.
Author information
Authors and Affiliations
Contributions
K.J.H.V. and J.L.T. conceived the manuscript and wrote the bulk of the initial draft. A.A. created the two figures, and J.M.V., A.A., N.A.G., and E.M.D. contributed to the writing of the manuscript. All authors critically revised the manuscript for important intellectual content.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Verweij, K.J.H., Vink, J.M., Abdellaoui, A. et al. The genetic aetiology of cannabis use: from twin models to genome-wide association studies and beyond. Transl Psychiatry 12, 489 (2022). https://doi.org/10.1038/s41398-022-02215-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-022-02215-2
