
Abstract
To date, little is known about the pleiotropic genetic variants among depression, cognition, and memory. The current research aimed to identify the potential pleiotropic single nucleotide polymorphisms (SNPs), genes, and pathways of the three phenotypes by conducting a multivariate genome-wide association study and an additional pleiotropy analysis among Chinese individuals and further validate the top variants in the UK Biobank (UKB). In the discovery phase, the participants were 139 pairs of dizygotic twins from the Qingdao Twins Registry. The genome-wide efficient mixed-model analysis identified 164 SNPs reaching suggestive significance (P < 1 × 10−5). Among them, rs3967317 (P = 1.21 × 10−8) exceeded the genome-wide significance level (P < 5 × 10−8) and was also demonstrated to be associated with depression and memory in pleiotropy analysis, followed by rs9863698, rs3967316, and rs9261381 (P = 7.80 × 10−8−5.68 × 10−7), which were associated with all three phenotypes. After imputation, a total of 457 SNPs reached suggestive significance. The top SNP chr6:24597173 was located in the KIAA0319 gene, which had biased expression in brain tissues. Genes and pathways related to metabolism, immunity, and neuronal systems demonstrated nominal significance (P < 0.05) in gene-based and pathway enrichment analyses. In the validation phase, 12 of the abovementioned SNPs reached the nominal significance level (P < 0.05) in the UKB. Among them, three SNPs were located in the KIAA0319 gene, and four SNPs were identified as significant expression quantitative trait loci in brain tissues. These findings may provide evidence for pleiotropic variants among depression, cognition, and memory and clues for further exploring the shared genetic pathogenesis of depression with Alzheimer’s disease.
Similar content being viewed by others

Introduction
Depression and Alzheimer’s disease (AD) are two common mental disorders that pose a serious threat to the physical and mental health of the public, especially elderly persons [1, 2], resulting in a heavy burden of disease worldwide [3, 4]. An increasing number of studies support that depression and AD are comorbidities [5, 6]. Evidence has shown some common pathogenic mechanisms and pathological changes between depression and AD, such as chronic inflammation [7, 8], dysregulation of the hypothalamic-pituitary-adrenal axis [9], lower levels of norepinephrine and 5-hydroxytryptamine [10], and a smaller volume of the hippocampus [11], suggesting the possibility and biological rationality of the basis of comorbidity between depression and AD.
In addition to environmental factors, genetic factors play an important etiologic role in both depression and AD. According to a meta-analysis of twin studies, the heritability of depression ranges from 31% to 42% [12]. As the most commonly used endophenotypes of AD in genetic studies [13, 14], the heritability of cognition and memory ranges from 47% to 59% and 36% to 47%, respectively, based on a large meta-analysis of twin and family studies [15]. Moreover, some evidence has suggested a shared genetic basis among depression, cognition, and memory. Pertinently, Franz et al. found that shared genetic effects could explain 77% of the correlation of early cognitive function with midlife depression in American male twins [16]. Another study in male twins showed that the genetic correlation of executive function with the genetic effects shared by persons with depression and anxiety was −0.44 [17]. The Colorado Adoption Project’s study revealed that the genetic correlations of memory with several dimensions of cognition ranged from 0.59 to 0.69 [18]. Additionally, our previous study utilizing multivariate twin models found that, among Qingdao twins, the genetic correlations were −0.31 for depression and cognition, −0.28 for depression and memory, and 0.69 for memory and cognition, respectively [19].
However, to date, little is known about the shared genetic variants among depression, cognition, and memory. A depression genome-wide meta-analysis identified 269 genes associated with depression [20]. Interestingly, 70 of the 269 depression-related genes were also found in another genome-wide meta-analysis of cognitive function [21]. In addition, the hNP gene was associated with both depression [22] and memory [23]. The APOE-4 allele gene was linked to lower cognitive ability, faster cognitive decline [24], and poorer memory [25]. Studies reported that DISC1 gene polymorphisms were associated with depression [26, 27], cognition [27], and memory [27, 28]. These overlapping findings from univariate studies provided indirect clues about the potentially shared susceptibility genes for depression, cognition, and memory, but there still has been a lack of direct evidence for the shared genetic variants and genes among the three phenotypes. Multivariate genome-wide association studies (GWASs) can be performed to search for potential pleiotropic genetic variants affecting multiple phenotypes by jointly modeling all phenotypes simultaneously rather than focusing on the simple overlap of genetic variants among different studies. Moreover, multivariate GWASs have higher statistical power and more accurate parameter estimation than univariate GWASs, which is helpful for the discovery of pleiotropic and small effects of genes [29, 30]. Notably, twins are particularly valuable for genetic studies owing to their sharing of rearing and intrauterine environments, as well as genetic similarity and discrepancy [31]. The combination of twin-based design with GWAS is excellent in controlling population stratification and ‘passive gene-environment correlation (rGE)’ and can distinguish direct genetic effects from indirect genetic effects [32]. Additionally, owing to the severe European bias of GWASs, it is unknown how many genetic risk loci can be translated across ethnicities [33]. Human genomics research among under-represented populations and cross-ethnic studies are urgently needed, given that cross-ethnicity generalizability is vital for improving genetic risk prediction and the applicability of therapeutic targets, alleviating bias and unfairness against specific subpopulations [34].
Thus, we performed a multivariate GWAS among Qingdao twins in China to explore the potential pleiotropic single nucleotide polymorphisms (SNPs), genes, and pathways among depression, cognition, and memory. An additional pleiotropy analysis was also performed for interpreting the possibility of pleiotropy. Then, to determine if these findings in Chinese can be generalized to different ethnic groups (cross-ethnicity generalizability), we further validated the top variants in an independent UK Biobank (UKB) population to identify cross-ethnic associations.
Materials and methods
Study population
In the discovery phase, the participants were adult twins from the Qingdao Twins Registry, China, and the details have been described in previous literature [19]. Blood samples were collected from participants after they fasted overnight, and identification of zygosity was carried out by sex, blood type, and microsatellite DNA gene scanning and typing. Participants who were monozygotic twins, were lactating or pregnant, had serious diseases or lacked biological sample information were excluded. Finally, the current multivariate GWAS sample included 139 dizygotic twin pairs.
Phenotypes
Depression was assessed using the 30-item Geriatric Depression Scale (GDS-30, Chinese version), which consists of 30 questions, with an overall score of 0-30 points, and a higher score indicated more severe depressive symptoms. The GDS-30 is especially suitable for the assessment of depression in middle-aged and elderly individuals and is also highly valid in the Chinese population [35, 36].
Cognition was measured using the Montreal Cognitive Assessment (MoCA, Chinese version) with high reliability and acceptance in Chinese adults [37, 38]. This assessment involved attention, naming, delayed recall, language, visuospatial/executive ability, orientation, and abstraction, with a total score of 30 points. To correct for the effect of education on cognitive performance, education-adjusted scores were used [39], where the scores of participants with ≤12 years of education were given one additional point, but with a total score of no more than 30. A lower cognition score indicated worse cognitive ability.
Memory was assessed by the backward and forward digit span tasks of the Wechsler Adult Intelligence (WAIS, Chinese version). The total score ranging from 0-17 was obtained by summing the scores of backward and forward digit span, and a lower score indicated worse memory. Digit span tasks have widely been used to reflect short-term memory, and the backward digit span also reflects working memory [40].
Genotyping, quality control, and imputation
Infinium Omni2.5Exome-8v1.2 BeadChip from Illumina was used for genotyping in dizygotic twins. After quality control, 1,338,905 SNPs with calling rate >0.98, locus missing <0.05, the significance of Hardy-Weinberg equilibrium (HWE) significance >1 × 10−4, and minor allele frequency (MAF) > 0.05 were included in this multivariate GWAS.
On the basis of the linkage disequilibrium (LD) principle, IMPUTE2 software [41] was utilized to impute untyped SNPs with reference to the data collected during the third phase of the 1000 Genomes Project (ASIAN) [42]. After filtering by HWE > 1 × 10−4, MAF > 0.05, and R2 > 0.6, a total of 7,399,084 SNPs were finally used in the post-imputation multivariate GWAS.
Multivariate GWAS
SNP-based analysis
The genome-wide efficient mixed-model association (GEMMA) [43] was used to evaluate the association of SNP genotypes with depression-cognition-memory phenotypic pairs after depression, cognition, and memory scores were transformed by rank transformation based on Blom’s formula [44] to normalize their skewed distributions, adjusting for sex, age, and the first five genetic principal components (PCs). The GEMMA fitted a multivariate linear mixed model (mvLMM) for testing marker associations with multiple phenotypes (depression, cognition, and memory) simultaneously while controlling for relatedness (here intra-pair correlation of twins) and population structure. The significance level was defined as a P value threshold of <5 × 10−8 (a conventional Bonferroni-corrected threshold) [45], and the suggestive level was defined as a P value threshold of <1 × 10−5 (a commonly utilized threshold in GWAS) [46]. Quantile-quantile (Q-Q) and Manhattan plots were used to visualize the results. Furthermore, enhancer enrichment analysis was performed by submitting the list of the top 100 SNPs (ranked by P values) associated with depression-cognition-memory to HaploReg v4.1 [47], and the cell-type enhancers with a P value of <0.05 were reported. All genomic coordinates were based on human genome Build 37 (NCBI GRCh37).
For interpreting the possibility of pleiotropy, we further performed a pleiotropy analysis by using the R package “pleio” [48] to test which phenotypes were associated with the potential pleiotropic genetic variants with suggestive significance in the current multivariate GWAS. Specifically, a new likelihood-ratio test with an extended sequential approach was used to test pleiotropy, which provides a testing framework to identify the number of phenotypes associated with a genetic variant, accounting for correlations among the phenotypes [48]. First, the sequential tests of pleiotropy started at the null hypothesis that all coefficients were equal to zero (test 0). If this test 0 was rejected, then test 1 was performed, which allowed one coefficient to be non-zero to test whether the remaining coefficients were equal to zero. If the test 1 was rejected, we then performed the test 2, which allowed two non-zero coefficients, considering all possible combinations of two non-zero coefficients and testing whether the remaining coefficients were equal to zero. Whenever a P value greater than 0.05 was derived, the sequential testing stopped. If the P value of test 2 remained <0.05, it implied that all three phenotypes were associated with this variant.
Gene-based analysis
VEGAS2 [49] was applied to carry out gene-based analysis by integrating the SNPs within a gene, and “1000 G East Asian Population” was used. A P value of <2.61 × 10−6 (0.05/19,152) was regarded as a significant threshold by Bonferroni correction due to the 19,152 genes tested, and the nominal significance level was defined as a P value of <0.05 [50].
Pathway enrichment analysis
PASCAL [51] was utilized to evaluate pathway scores. SNPs were mapped to genes, and then the joint score of all genes involved in a pathway was calculated. The chi-squared and empirical scores were utilized to assess pathway enrichment of high-scoring genes. The Reactome, KEGG, and BioCarta databases were used to obtain pathway information. An emp-P value of <4.64 × 10−5 (0.05/1,077) was regarded as a significant threshold by Bonferroni correction due to the 1,077 pathways tested, and the nominal significance level was defined as an emp-P value of <0.05.
Validation analysis
To identify the cross-ethnicity generalizability and cross-ethnic associations, we validated the top variants in an independent UKB population, which is a population-based cohort of 488,377 individuals with genotypic data across the United Kingdom; more details of genotyping, quality control, and imputation have been described elsewhere [52]. The phenotypic and genotypic data utilized in the current study were obtained from the third version of UKB data under an approved data application (application number: 66354). Depression was assessed using the two-item Patient Health Questionnaire (PHQ-2), and a total score of three and more indicated possible depression [53]. Cognition was measured through 13 numerical and verbal reasoning questions reflecting reasoning ability, and correct scores ranged from 0–13. Memory was assessed by the digit span task, and the maximum digits remembered correctly ranged from 2–12. A total of 46,102 individuals participated in and completed the depression, cognition, and memory tests. Cases (n = 355) were defined as participants with depression scores ≥3 and cognition and memory performance scores lower than the 25th percentile of their score distributions. Controls (n = 1775) were selected by matching individuals’ age and sex (ratio = 1:5) with those of the participants (n = 30,470) with depression scores <3 and cognition and memory performance ≥25th percentile of their score distributions. Finally, 2130 individuals (355 cases and 1775 controls) with a median age (interquartile range) of 55 (15) years were included in the validated sample. The top SNPs were validated by logistic regression analysis of the additive effect model, adjusting for the first 10 genetic PCs. A total of 469 of 481 SNPs (the union number before and after imputation) with P values lower than 1 × 10−5 in the discovery set were typed in the UKB data set and selected for validation. Thus, a P value < 1.07 × 10−4 (0.05/469) was regarded as a significant threshold by Bonferroni correction, and the nominal significance level was defined as a P value <0.05. Statistical analyses were performed utilizing R version 4.1.0.
Expression quantitative trait loci (eQTL) analysis
For SNPs with nominal significance in the validation set, we further checked their functional consequences by eQTL analysis across tissues using data from the GTEx portal (version 8) [54]. A P value lower than 0.05 was regarded as significant in the single-tissue eQTL analysis. The posterior probability m-value that the eQTL effect existed in each tissue of a cross-tissue meta-analysis higher than 0.9 indicated that the tissue had an eQTL effect [55]. An outline of the overall study design and analysis steps is shown in Fig. 1.

Flowchart of the overall study design and analysis steps.
Results
Basic characteristics
There were 139 pairs of dizygotic twins in the final discovery sample. The median (interquartile range) age was 49 (11) years, and the median scores (interquartile ranges) for depression, cognition, and memory for participants were 7 (7), 22 (5), and 12 (3) points, respectively (Supplementary Table 1).
Multivariate GWAS
SNP-based analysis
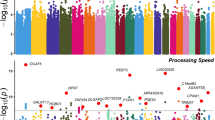
In the SNP-based study, the Q-Q plot (Fig. 2a) suggested no evidence of population stratification. The Manhattan plot (Fig. 3a) demonstrated that a total of 164 SNPs reached the level of suggestive significance (P < 1 × 10−5) (Supplementary Table 2); among them, rs3967317 (P = 1.21 × 10−8) on the CNTN4 gene on chromosome 3 exceeded the genome-wide significance level (P < 5 × 10−8). In addition, rs9863698 (P = 7.80 × 10−8) and rs3967316 (P = 1.33 × 10−7) on the CNTN4 gene, rs9261381 (P = 5.68 × 10−7) on the TRIM31 gene, rs11577464 (P = 5.82 × 10−7) on the LINC02567 gene, and rs73198369 (P = 7.00 × 10−7) on the RNU6-1325P gene reached suggestive significance. The top 20 SNPs ranked by P values are shown in Table 1. The additional pleiotropy analysis identified that 144 of the 164 SNPs (87.8%) were associated with two or three phenotypes (P < 0.05), with rs3967317 associated with depression and memory, rs9863698, rs3967316, and rs9261381 associated with all three phenotypes (Supplementary Table 2).

a The quantile-quantile plot based on data before imputation. b The quantile-quantile plot based on data after imputation. The horizontal axis represents the expected −log10 (P), while the vertical axis represents the observed −log10 (P). The red line represents the expectation of the null hypothesis of no association, and the gray shaded area represents 95% confidence intervals of the null hypothesis. The black dots represent the observed data, and λ indicates genomic inflation.

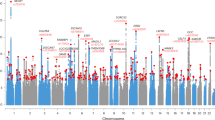
a The Manhattan plot based on data before imputation. b The Manhattan plot based on data after imputation. The horizontal axis represents autosomes and the X chromosome, while the vertical axis represents the P values of SNPs. The red line represents the genome-wide significance threshold (5 × 10−8), and the lower horizontal dashed line represents the suggestive significance level (1 × 10−5).
Enhancer enrichment analysis found that the top 100 depression-cognition-memory-related genetic variants were significantly enriched in six tissues and cells, including pancreatic islets, stomach mucosa, fetal intestine large, primary natural killer cells, and T regulatory cells from peripheral blood, and liver (P < 0.05) (Supplementary Table 3).
After using the data of the third phase of the 1000 Genomes Project as a reference to impute untyped SNPs, there was still no evidence of population stratification (Fig. 2b), and the amounts of SNPs with suggestive significance increased, with a total of 457 SNPs reaching the level of suggestive significance (Fig. 3b, Supplementary Table 4). The first three top SNPs, chr6:24597173, rs12210323, and rs12213116 (P = 1.71 × 10−7−1.72 × 10−7), were located in the KIAA0319 gene, which had biased expression in multiple brain tissues (Supplementary Fig 1), followed by rs61783213 (P = 2.77 × 10−7) in the LINC02567 gene. The top 20 SNPs after imputation are shown in Supplementary Table 5. A total of 377 of the 457 SNPs (82.5%) were associated with two or three phenotypes (P < 0.05) (Supplementary Table 4).
Gene-based analysis
In the gene-based study, no statistically significant gene was found (P < 2.61 × 10−6), but 1107 genes reached the nominal significance level (P < 0.05). Most of these genes are known to be involved in metabolism, immunity, and neuronal systems, and the top 20 genes are shown in Table 2.
Pathway enrichment analysis
In the pathway-based analysis, no statistically significant pathway was found (P < 4.64 × 10−5), but 587 pathways were found to be nominally associated with depression-cognition-memory (emp-P < 0.05), and most of these pathways were involved in the metabolism of amino acids, lipids and RNA, the immune system, and the neuronal system. The top 20 pathways are shown in Table 3.
Validation analysis
A total of 469 SNPs with P values lower than 1 × 10−5 in the discovery set had genotype data in the UKB validation set and were selected for validation. Although no SNP passed the Bonferroni correction level, 12 SNPs reached the nominal significance level (P < 0.05), three of them (rs13209442, rs13208577, and rs12213116) were located in the KIAA0319 gene, and one (rs9261134) was located in the ZNRD1ASP gene (Supplementary Table 6).
eQTL analysis
The eQTL analysis across tissues found that four (rs2539731, rs17337582, rs62358383, and rs9261134) of 12 SNPs with nominal significance in the validation set were significant eQTLs in multiple tissues, specifically in brain tissues (Supplementary Figs 2–5). Among these SNPs, rs2539731 (Supplementary Fig 2, brain-substantia nigra: P = 1.10 × 10−3, m-value = 0.965; brain-nucleus accumbens: P = 3.60 × 10−5, m-value = 0.991), rs17337582 (Supplementary Fig 3, brain-substantia nigra: P = 1.20 × 10−3, m-value = 0.951; brain-nucleus accumbens: P = 4.10 × 10−5, m-value=0.996), and rs62358383 (Supplementary Fig 4, brain-substantia nigra: P = 1.90 × 10−3, m-value = 0.927; brain-nucleus accumbens: P = 5.70 × 10−5, m-value = 0.987) were significantly associated with the expression of MAP3K1 gene in brain tissues. Rs9261134 located at the ZNRD1ASP gene was significantly associated with the expression of 15 genes (Supplementary Table 6) in multiple brain tissues (brain-nucleus accumbens, brain-frontal cortex, brain-caudate, brain-putamen, brain-hypothalamus, brain-amygdala, brain-cortex, and brain-anterior cingulate cortex) (P < 0.5, m-value > 0.9), and ZNRD1ASP expression with rs9261134 across tissues is shown in Supplementary Fig 5.
Discussion
The current study performed the first multivariate GWAS of depression-cognition-memory and found some pleiotropic SNPs, genes, and pathways among depression, cognition, and memory in Qingdao twins. Moreover, multiple variants were replicated in another independent UKB population.
Although few previous achievements have been made involving pleiotropic variants of depression, cognition, and memory across ancestries, some recent literature has focused on both depression and cognitive function or depression related variants across ancestry groups. Thalamuthu et al. performed a genome-wide interaction analysis of major depressive disorder (MDD) with cognitive function among European cohorts. The study revealed that MDD status had a moderating effect on the associations of variants with cognitive function, with some SNPs associated with cognitive domains in the context of MDD [56]. Another study conducted pleiotropy analyses, utilizing MDD and late-onset AD GWAS data based on European ancestry, thereby indicating that the genetic risks associated with AD might influence MDD risk [57]. Cross-ethnic studies, similar to our findings, demonstrated a small shared polygenic basis for depression in European and East Asian populations. Bigdeli et al. found a weak overlap of SNP effects between East Asian and European ancestries by combining MDD GWAS summary statistics of Chinese and European participants [58]. One significant SNP (rs10912903) was replicated in the current multivariate GWAS, with a P value of 7.15 × 10−3. Another study also showed that only 11% of depression risk loci previously identified in the European population reached nominal significance in the East Asian population [59].
In the SNP-based analysis, the strongest association signal was rs3967317 located in the CNTN4 gene on chromosome 3, which exceeded the genome-wide significance level. The following were rs9863698 and rs3967316, which were also located in the CNTN4 gene. The CNTN4-encoded protein belongs to the contactin family and is involved in neuronal network development and plasticity. Pertinently, studies have found that CNTN4 is associated with mental retardation [60] and affects intelligence [61]. Rs9261381 was located in the TRIM31 gene on chromosome 6. TRIM31 encodes a protein that functions as an E3 ubiquitin-protein ligase and can regulate cell growth. Studies have shown that the TRIM31 gene was associated with intelligence only in the background of a psychiatric disorder [62]. Furthermore, the top 100 depression-cognition-memory related genetic variants were significantly enriched in pancreatic islets, stomach mucosa, fetal intestine large, primary natural killer cells and T regulatory cells from peripheral blood, and the liver. Ample evidence has shown that these tissues and cells are closely related to depression, cognition, and memory [63,64,65,66,67,68,69,70], which further supports our findings.
After imputing untyped SNPs, more SNPs with suggestive significance were identified. The SNP chr6:24597173 located in the KLAA0319 gene showed the strongest association, although this might be mainly driven by the association of the SNP with cognition. However, the KLAA0319 gene demonstrated a biased expression in multiple brain tissues, and three of 12 SNPs with nominal significance in the validation phase among the UKB population whose cases were abnormal on all three phenotypes and controls were normal were located in the KIAA0319 gene, indicating a potential cross-ethnic association of KIAA0319 with depression-cognition-memory. The protein encoded by KIAA0319 can regulate cell adhesion and neuronal migration processes to influence the growth of the cerebral cortex, and KIAA0319 has been found to be associated with category fluency, recall process, and verbal learning [71]. In addition, four of the other nine SNPs were significant eQTLs in brain tissues that control mood, cognition, and memory; moreover, three SNPs were significantly associated with the expression level of the MAP3K1 gene in the brain-substantia nigra and brain-nucleus accumbens. Pertinently, the protein encoded by MAP3K1 is part of the nuclear factor kappa beta (NF-κB) pathway [72]. NF-κB has been found to be involved in the pathophysiology of depression [73] and is closely related to the pathogenesis of AD [74].
Most genes with nominal significance levels in the gene-based analysis are known to be involved in metabolism, immunity, and neuronal systems. The potential mechanisms of several interesting genes other than those mentioned above were as follows: (1) MEDAG is an adipogenic gene that can promote the formation of adipocytes [75]. The lipid metabolism process involves the pathogenesis of depression and AD [76, 77]; (2) the C3a peptide encoded by C3 can modulate the inflammation process which is closely related to depression and AD [7, 8]; and (3) the protein encoded by TET1 influences gene activation and the process of DNA methylation. Studies have shown that the expression level of TET1 is significantly increased in psychotic participants [78], and TET1 variation is associated with late-onset AD [79]; (4) FGF1 is related to the survival of neurons and is involved in various biological processes. Furthermore, FGF1 has been reported to be associated with AD in Han Chinese individuals [80].
In the pathway enrichment analysis, most pathways that reached nominal significance were related to metabolism, immune, and neuronal system, and several important pathways were revealed as follows: (1) metabolism of amino acids and derivatives, various metabolites of this pathway including glutamate and glycine are involved in the pathophysiology of depression and AD [81, 82]; (2) sphingolipid de novo biosynthesis and (3) sphingolipid metabolism pathways, both of which are involved in the metabolism process of sphingolipid. The intermediate product is ceramide, which is closely related to the pathological mechanisms of depression and AD and used as a therapeutic target [83]. For (4) N-glycan antennae elongation in the medial/trans-Golgi and (5) N-glycan antennae elongation, there are significant differences between depression patients and controls in the serum N-glycan structure levels [84], and N-glycans can influence the development and progression of AD by regulating the key glycoproteins [85]. (6) Immunoregulatory interactions between a lymphoid and a non-lymphoid cell, which is an important pathway in the immune system; relevantly, the immune system is regarded as a major factor in both depression and AD [86, 87]. A previous study also found immune-related pathways in the shared genetic etiology of depression and AD [57]. (7) Other semaphorin interactions pathway containing four types of plexins and eight classes of semaphorins is involved in axon guidance and the development of the nervous system [88]. Semaphorins have been related to major depression risk [89], and plexin-A4 can mediate Aβ-induced tau pathology in the pathogenesis of AD [90]. However, some pathways, such as cortisol/stress responses and cholinergic and serotonergic function [10], have been clearly linked to both depression and AD but were not identified as top pathways in the current study with higher P values (stress pathway: emp-P = 2.60 × 10−2, neurotransmitter release cycle: emp-P = 2.16 × 10−2, acetylcholine neurotransmitter release cycle pathway: emp-P = 2.19 × 10−2, etc.). Except for the limitation of the small sample size, another possible reason was that although several physiological processes were thought to be the common mechanisms of both depression and AD, they might be mainly caused by different genes in depression and AD, rather than pleiotropic genes, and further research is required in the future.
The current multivariate GWAS had several strengths. First, this is the first study to identify potential pleiotropic SNPs, genes, and pathways among depression, cognition, and memory phenotypes in Chinese individuals, which may not only provide insight into a common genetic basis of these phenotypes but also make a little contribution in shifting the Eurocentric bias of GWASs. Second, this current GWAS was performed in twin samples. The twin-based GWAS design has been demonstrated excellence in controlling population stratification and passive rGE and can identify direct genetic effects [32], which reduced the concerns about false-positive errors and the confusion of indirect genetic effects. Third, validation analysis was performed in another independent UKB population, which allowed the identification of cross-ethnic generalizability. However, several study limitations should also be considered. First, the sample size of the current study was relatively small owing to the difficulty in twin pair recruitment, even though the algorithm of multivariate GWAS has the natural advantage of power [91], which may restrict the discovery of more significantly associated SNPs, genes, and pathways. In fact, no gene and pathway reached the significance threshold of Bonferroni correction, which might be partly due to the small sample size, and further research in a large East Asian population is needed. Notably, cognitive decline in depressed individuals could be secondary to depression itself, such as psychomotor slowing and withdrawal from engagement in activities that were conducive to cognition. Similarly, cognitive dysfunction might lead to stress and psychological burdens that give rise to depressive symptoms. In these cases, the genetics affecting cognition and memory might largely be separate from those that served as other risk factors for depression, which might also explain the lack of extensive genetic overlap among the three phenotypes in the current study. Second, the assessment methods of depression, cognition, and memory phenotypes were not perfectly consistent between the discovery set and validation set; for example, depression was assessed using the GDS-30 in the current multivariate GWAS and the PHQ-2 in UKB validation analysis. Although both the GDS-30 and PHQ-2 are reliable and valid and have been widely used as screening measures for depression, there may still be phenotypic heterogeneity to some degree due to the not identical questionnaire items. The phenotypic heterogeneity and ethnic difference in the MAF and LD structure might partly account for the fewer significant findings revealed in the validation analysis. Nevertheless, we still found a potential cross-ethnic association of KIAA0319. However, the strongest SNP (rs3967317), which demonstrated genome-wide significance (P < 5 × 10−8) and still showed suggestive significance after imputation (P < 1 × 10−5) in the discovery set, was not replicated in the UKB validation sample, but it may still be worth being further validated in the Asian population as a promising candidate variant.
In conclusion, this multivariate GWAS identified some pleiotropic SNPs, genes, and pathways among depression, cognition, and memory, which provided evidence for a common genetic basis of the three phenotypes and clues for further exploring the shared genetic pathogenesis of depression with AD, and it might be helpful in the search for new therapeutic targets for both diseases.
Data availability
The dataset used in the present study is available from the corresponding author upon reasonable request.
References
Assariparambil AR, Noronha JA, Kamath A, Adhikari P, Nayak BS, Shankar R, et al. Depression among older adults: a systematic review of South Asian countries. Psychogeriatrics 2021;21:201–19.
Zhang Y, Li Y, Ma L. Recent advances in research on Alzheimer’s disease in China. J Clin Neurosci. 2020;81:43–46.
WORLD HEALTH ORGANIZATION. World Federation for Mental Health. DEPRESSION: A Global Crisis. World Mental Health Day, October 10 2012.2012: https://www.who.int/mental_health/management/depression/wfmh_paper_depression_wmhd_2012.pdf. Accessed 18 April 2021.
Hodson R. Alzheimer’s disease. Nature 2018;559:S1.
Wang JH, Wu YJ, Tee BL, Lo RY. Medical comorbidity in Alzheimer’s disease: a nested case-control study. J Alzheimers Dis. 2018;63:773–81.
Starkstein SE, Jorge R, Mizrahi R, Robinson RG. The construct of minor and major depression in Alzheimer’s disease. Am J Psychiatry. 2005;162:2086–93.
Wohleb ES, Franklin T, Iwata M, Duman RS. Integrating neuroimmune systems in the neurobiology of depression. Nat Rev Neurosci. 2016;17:497–511.
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421.
Caraci F, Copani A, Nicoletti F, Drago F. Depression and Alzheimer’s disease: neurobiological links and common pharmacological targets. Eur J Pharm. 2010;626:64–71.
Hao W, Yu X Psychiatry. 7th ed. Beijing: People’s Medical Publishing House; 2013.
Geuze E, Vermetten E, Bremner JD. MR-based in vivo hippocampal volumetrics: 2. Find Neuropsychiatr Disord Mol Psychiatry. 2005;10:160–84.
Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000;157:1552–62.
Seshadri S, DeStefano AL, Au R, Massaro JM, Beiser AS, Kelly-Hayes M, et al. Genetic correlates of brain aging on MRI and cognitive test measures: a genome-wide association and linkage analysis in the Framingham Study. BMC Med Genet. 2007;8:S15.
Reitz C, Mayeux R. Endophenotypes in normal brain morphology and Alzheimer’s disease: a review. Neuroscience 2009;164:174–90.
Blokland GAM, Mesholam-Gately RI, Toulopoulou T, Del Re EC, Lam M, DeLisi LE, et al. Heritability of Neuropsychological Measures in Schizophrenia and Nonpsychiatric Populations: A Systematic Review and Meta-analysis. Schizophr Bull. 2017;43:788–800.
Franz CE, Lyons MJ, O’Brien R, Panizzon MS, Kim K, Bhat R, et al. A 35-year longitudinal assessment of cognition and midlife depression symptoms: the Vietnam Era Twin Study of Aging. Am J Geriatr Psychiatry. 2011;19:559–70.
Gustavson DE, Franz CE, Panizzon MS, Reynolds CA, Xian H, Jacobson KC, et al. Genetic and environmental associations among executive functions, trait anxiety, and depression symptoms in middle age. Clin Psychol Sci. 2019;7:127–42.
Alarcón M, Plomin R, Fulker DW, Corley R, DeFries JC. Multivariate path analysis of specific cognitive abilities data at 12 years of age in the Colorado Adoption Project. Behav Genet. 1998;28:255–64.
Xu C, Sun J, Ji F, Tian X, Duan H, Zhai Y, et al. The genetic basis for cognitive ability, memory, and depression symptomatology in middle-aged and elderly chinese twins. Twin Res Hum Genet. 2015;18:79–85.
Howard DM, Adams MJ, Clarke TK, Hafferty JD, Gibson J, Shirali M, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019;22:343–52.
Davies G, Lam M, Harris SE, Trampush JW, Luciano M, Hill WD, et al. Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat Commun. 2018;9:2098.
Bobińska K, Mossakowska-Wójcik J, Szemraj J, Gałecki P, Zajączkowska M, Talarowska M. Human neuropsin gene in depression. Psychiatr Danub. 2017;29:195–200.
Izumi A, Iijima Y, Noguchi H, Numakawa T, Okada T, Hori H, et al. Genetic variations of human neuropsin gene and psychiatric disorders: polymorphism screening and possible association with bipolar disorder and cognitive functions. Neuropsychopharmacology 2008;33:3237–45.
Lipnicki DM, Makkar SR, Crawford JD, Thalamuthu A, Kochan NA, Lima-Costa MF, et al. Determinants of cognitive performance and decline in 20 diverse ethno-regional groups: A COSMIC collaboration cohort study. PLoS Med. 2019;16:e1002853.
Small GW, Chen ST, Komo S, Ercoli L, Bookheimer S, Miller K, et al. Memory self-appraisal in middle-aged and older adults with the apolipoprotein E-4 allele. Am J Psychiatry. 1999;156:1035–8.
Thomson PA, Parla JS, McRae AF, Kramer M, Ramakrishnan K, Yao J, et al. 708 Common and 2010 rare DISC1 locus variants identified in 1542 subjects: analysis for association with psychiatric disorder and cognitive traits. Mol Psychiatry. 2014;19:668–75.
Carless MA, Glahn DC, Johnson MP, Curran JE, Bozaoglu K, Dyer TD, et al. Impact of DISC1 variation on neuroanatomical and neurocognitive phenotypes. Mol Psychiatry. 2011;16:1096–104. 1063
Porteous DJ, Thomson P, Brandon NJ, Millar JK. The genetics and biology of DISC1-an emerging role in psychosis and cognition. Biol Psychiatry. 2006;60:123–31.
Zhang L, Pei YF, Li J, Papasian CJ, Deng HW. Univariate/multivariate genome-wide association scans using data from families and unrelated samples. PLoS One. 2009;4:e6502.
Allison DB, Thiel B, St Jean P, Elston RC, Infante MC, Schork NJ. Multiple phenotype modeling in gene-mapping studies of quantitative traits: power advantages. Am J Hum Genet. 1998;63:1190–201.
Neale MC, Cardon LR, Methodology for genetic studies of twins and families: Dordrecht: Kluwer Academic Publisher; 1992.
Friedman NP, Banich MT, Keller MC. Twin studies to GWAS: there and back again. Trends Cogn Sci. 2021;25:855–69.
Hindorff LA, Bonham VL, Brody LC, Ginoza MEC, Hutter CM, Manolio TA, et al. Prioritizing diversity in human genomics research. Nat Rev Genet. 2018;19:175–85.
Fu J, Festen EA, Wijmenga C. Multi-ethnic studies in complex traits. Hum Mol Genet. 2011;20:R206–213.
Yesavage JA, Brink TL, Rose TL, Lum O, Huang V, Adey M, et al. Development and validation of a geriatric depression screening scale: a preliminary report. J Psychiatr Res. 1982;17:37–49.
Niu L, Jia C, Ma Z, Wang G, Yu Z, Zhou L. Validating the Geriatric Depression Scale with proxy-based data: A case-control psychological autopsy study in rural China. J Affect Disord. 2018;241:533–8.
Nasreddine ZS, Phillips NA, Bédirian V, Charbonneau S, Whitehead V, Collin I, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53:695–9.
Chen KL, Xu Y, Chu AQ, Ding D, Liang XN, Nasreddine ZS, et al. Validation of the Chinese version of montreal cognitive assessment basic for screening mild cognitive impairment. J Am Geriatr Soc. 2016;64:e285–e290.
Peavy GM, Jenkins CW, Little EA, Gigliotti C, Calcetas A, Edland SD, et al. Community memory screening as a strategy for recruiting older adults into Alzheimer’s disease research. Alzheimers Res Ther. 2020;12:78.
Hubbard L, Tansey KE, Rai D, Jones P, Ripke S, Chambert KD, et al. Evidence of Common genetic overlap between Schizophrenia and cognition. Schizophr Bull. 2016;42:832–42.
Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–13.
Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, et al. A global reference for human genetic variation. Nature 2015;526:68–74.
Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44:821–4.
Blom G, Statistical estimates and transformed beta-variables. New York: John Wiley & Sons, Inc.; 1958.
Dudbridge F, Gusnanto A. Estimation of significance thresholds for genomewide association scans. Genet Epidemiol. 2008;32:227–34.
Loukola A, Wedenoja J, Keskitalo-Vuokko K, Broms U, Korhonen T, Ripatti S, et al. Genome-wide association study on detailed profiles of smoking behavior and nicotine dependence in a twin sample. Mol Psychiatry. 2014;19:615–24.
Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016;44:D877–881.
Schaid DJ, Tong X, Larrabee B, Kennedy RB, Poland GA, Sinnwell JP. Statistical methods for testing genetic pleiotropy. Genetics 2016;204:483–97.
Mishra A, Macgregor S. VEGAS2: Software for more flexible gene-based testing. Twin Res Hum Genet. 2015;18:86–91.
Xu C, Zhang D, Wu Y, Tian X, Pang Z, Li S, et al. A genome-wide association study of cognitive function in Chinese adult twins. Biogerontology 2017;18:811–9.
Lamparter D, Marbach D, Rueedi R, Kutalik Z, Bergmann S. Fast and rigorous computation of gene and pathway scores from SNP-based summary statistics. PLoS Comput Biol. 2016;12:e1004714.
Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018;562:203–9.
Kroenke K, Spitzer RL, Williams JB. The Patient Health Questionnaire-2: validity of a two-item depression screener. Med Care. 2003;41:1284–92.
The Genotype-Tissue Expression (GTEx) project. Nat Genet. 45, 580−5 (2013).
Han B, Eskin E. Interpreting meta-analyses of genome-wide association studies. PLoS Genet. 2012;8:e1002555.
Thalamuthu A, Mills NT, Berger K, Minnerup H, Grotegerd D, Dannlowski U, et al. Genome-wide interaction study with major depression identifies novel variants associated with cognitive function. Mol Psychiatry. 2021.
Lutz MW, Sprague D, Barrera J, Chiba-Falek O. Shared genetic etiology underlying Alzheimer’s disease and major depressive disorder. Transl Psychiatry. 2020;10:88.
Bigdeli TB, Ripke S, Peterson RE, Trzaskowski M, Bacanu SA, Abdellaoui A, et al. Genetic effects influencing risk for major depressive disorder in China and Europe. Transl Psychiatry. 2017;7:e1074.
Giannakopoulou O, Lin K, Meng X, Su MH, Kuo PH, Peterson RE, et al. The genetic architecture of depression in individuals of East Asian ancestry: a genome-wide association study. JAMA Psychiatry. 2021;78:1258–69.
Dijkhuizen T, van Essen T, van der Vlies P, Verheij JB, Sikkema-Raddatz B, van der Veen AY, et al. FISH and array-CGH analysis of a complex chromosome 3 aberration suggests that loss of CNTN4 and CRBN contributes to mental retardation in 3pter deletions. Am J Med Genet A. 2006;140:2482–7.
Loo SK, Shtir C, Doyle AE, Mick E, McGough JJ, McCracken J, et al. Genome-wide association study of intelligence: additive effects of novel brain expressed genes. J Am Acad Child Adolesc Psychiatry. 2012;51:432–.e432.
Rizzi TS, Arias-Vasquez A, Rommelse N, Kuntsi J, Anney R, Asherson P, et al. The ATXN1 and TRIM31 genes are related to intelligence in an ADHD background: evidence from a large collaborative study totaling 4,963 subjects. Am J Med Genet B Neuropsychiatr Genet. 2011;156:145–57.
Hung YJ, Hsieh CH, Chen YJ, Pei D, Kuo SW, Shen DC, et al. Insulin sensitivity, proinflammatory markers and adiponectin in young males with different subtypes of depressive disorder. Clin Endocrinol. 2007;67:784–9.
Nicolls MR. The clinical and biological relationship between Type II diabetes mellitus and Alzheimer’s disease. Curr Alzheimer Res. 2004;1:47–54.
Mayer EA, Craske M, Naliboff BD. Depression, anxiety, and the gastrointestinal system. J Clin Psychiatry. 2001;62:28–36.
de JRD-PV, Forlenza AS, Forlenza OV. Relevance of gutmicrobiota in cognition, behaviour and Alzheimer’s disease. Pharm Res. 2018;136:29–34.
Irwin M, Clark C, Kennedy B, Christian Gillin J, Ziegler M. Nocturnal catecholamines and immune function in insomniacs, depressed patients, and control subjects. Brain Behav Immun. 2003;17:365–72.
Jiang Q, Jin S, Jiang Y, Liao M, Feng R, Zhang L, et al. Alzheimer’s disease variants with the genome-wide significance are significantly enriched in immune pathways and active in immune cells. Mol Neurobiol. 2017;54:594–600.
Mullish BH, Kabir MS, Thursz MR, Dhar A. Review article: depression and the use of antidepressants in patients with chronic liver disease or liver transplantation. Aliment Pharm Ther. 2014;40:880–92.
Weinstein G, Davis-Plourde K, Himali JJ, Zelber-Sagi S, Beiser AS, Seshadri S. Non-alcoholic fatty liver disease, liver fibrosis score and cognitive function in middle-aged adults: The Framingham Study. Liver Int. 2019;39:1713–21.
Nicodemus KK, Elvevåg B, Foltz PW, Rosenstein M, Diaz-Asper C, Weinberger DR. Category fluency, latent semantic analysis and schizophrenia: a candidate gene approach. Cortex 2014;55:182–91.
Schichl YM, Resch U, Lemberger CE, Stichlberger D, de Martin R. Novel phosphorylation-dependent ubiquitination of tristetraprolin by mitogen-activated protein kinase/extracellular signal-regulated kinase kinase kinase 1 (MEKK1) and tumor necrosis factor receptor-associated factor 2 (TRAF2). J Biol Chem. 2011;286:38466–77.
Caviedes A, Lafourcade C, Soto C, Wyneken U. BDNF/NF-κB signaling in the neurobiology of depression. Curr Pharm Des. 2017;23:3154–63.
Ju Hwang C, Choi DY, Park MH, Hong JT. NF-κB as a key mediator of brain inflammation in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2019;18:3–10.
Zhang H, Chen X, Sairam MR. Novel genes of visceral adiposity: identification of mouse and human mesenteric estrogen-dependent adipose (MEDA)-4 gene and its adipogenic function. Endocrinology 2012;153:2665–76.
Maes M, Smith R, Christophe A, Vandoolaeghe E, Van Gastel A, Neels H, et al. Lower serum high-density lipoprotein cholesterol (HDL-C) in major depression and in depressed men with serious suicidal attempts: relationship with immune-inflammatory markers. Acta Psychiatr Scand. 1997;95:212–21.
Zarrouk A, Debbabi M, Bezine M, Karym EM, Badreddine A, Rouaud O, et al. Lipid biomarkers in Alzheimer’s disease. Curr Alzheimer Res. 2018;15:303–12.
Dong E, Gavin DP, Chen Y, Davis J. Upregulation of TET1 and downregulation of APOBEC3A and APOBEC3C in the parietal cortex of psychotic patients. Transl Psychiatry. 2012;2:e159.
Morgan AR, Hamilton G, Turic D, Jehu L, Harold D, Abraham R, et al. Association analysis of 528 intra-genic SNPs in a region of chromosome 10 linked to late onset Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet. 2008;147b:727–31.
Tao QQ, Sun YM, Liu ZJ, Ni W, Yang P, Li HL, et al. A variant within FGF1 is associated with Alzheimer’s disease in the Han Chinese population. Am J Med Genet B Neuropsychiatr Genet. 2014;165b:131–6.
Zhang Y, Yuan S, Pu J, Yang L, Zhou X, Liu L, et al. Integrated metabolomics and proteomics analysis of hippocampus in a rat model of depression. Neuroscience 2018;371:207–20.
Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem Int. 2004;45:583–95.
Brodowicz J, Przegaliński E, Müller CP, Filip M. Ceramide and its related neurochemical networks as targets for some brain disorder therapies. Neurotox Res. 2018;33:474–84.
Boeck C, Pfister S, Bürkle A, Vanhooren V, Libert C, Salinas-Manrique J, et al. Alterations of the serum N-glycan profile in female patients with Major Depressive Disorder. J Affect Disord. 2018;234:139–47.
Kizuka Y, Kitazume S. Taniguchi N. N-glycan and Alzheimer’s disease. Biochim Biophys Acta Gen Subj. 2017;1861:2447–54.
Yrondi A, Sporer M, Péran P, Schmitt L, Arbus C, Sauvaget A. Electroconvulsive therapy, depression, the immune system and inflammation: A systematic review. Brain Stimul. 2018;11:29–51.
Jevtic S, Sengar AS, Salter MW, McLaurin J. The role of the immune system in Alzheimer disease: Etiology and treatment. Ageing Res Rev. 2017;40:84–94.
Gil V, Del Río JA, Functions of plexins/neuropilins and their ligands during hippocampal development and neurodegeneration. Cells. 8, (2019).
Zhou H, Polimanti R, Yang BZ, Wang Q, Han S, Sherva R, et al. Genetic risk variants associated with comorbid alcohol dependence and major depression. JAMA Psychiatry. 2017;74:1234–41.
Chung S, Yang J, Kim HJ, Hwang EM, Lee W, Suh K, et al. Plexin-A4 mediates amyloid-β-induced tau pathology in Alzheimer’s disease animal model. Prog Neurobiol. 2021;203:102075.
Zhou X, Stephens M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat Methods. 2014;11:407–9.
Acknowledgements
We thank Dr. Gu Zhu for his technical guidance of data analysis. We thank all participants and contributors of Qingdao Twins and the UK Biobank. This study was supported by the National Natural Science Foundation of China (82073641).
Author information
Authors and Affiliations
Contributions
JS, DZ, and XL designed the study. HD, XT, and CX collected samples and phenotypes. JS, RZ, XL, and DZ contributed to data analysis and interpretation of results. JS drafted the manuscript. DZ, XL, and WW reviewed and revised the manuscript. All authors reviewed and commented on subsequent drafts of the manuscript. All authors read and approved the final manuscript as submitted and published.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was conducted with adherence to the Declaration of Helsinki and was approved by the Regional Ethics Committee of the Qingdao Center for Disease Control and Prevention and National Health Service Research Ethics Committee. All participants provided informed consent.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, J., Wang, W., Zhang, R. et al. Multivariate genome-wide association study of depression, cognition, and memory phenotypes and validation analysis identify 12 cross-ethnic variants. Transl Psychiatry 12, 304 (2022). https://doi.org/10.1038/s41398-022-02074-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-022-02074-x
