
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive and lethal malignancy characterized by dysregulated energy and stromal metabolism. It is strongly supported by activated pancreatic stellate cells (PSC) which drive excessive desmoplasia and tumor growth via metabolic crosstalk. Herein, a programmed nanosystem is designed to dual rectify the metabolism abnormalities of the PDAC cells, which overexpress glucose transporter 1(GLUT1) and CD71, and the PSC for oncotherapy. The nanosystem is based on a tumor microenvironment-responsive liposome encapsulating an NF-κB inhibitor (TPCA-1) and a CD71 aptamer-linked Glut1 siRNA. TPCA-1 reverses the activated PSC to quiescence, which hampers metabolic support of the PSC to PDAC cells and bolsters the PDAC cell-targeting delivery of the siRNA. Aerobic glycolysis and the following enhancement of oxidative phosphorylation are restrained by the nano-modulation so as to amplify anti-PDAC efficacy in an orthotopic xenograft mouse model, which implies more personalized PDAC treatment based on different energy metabolic profiles.
Similar content being viewed by others

Introduction
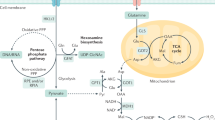
Pancreatic ductal adenocarcinoma (PDAC) is predicted to become the third primary cause of cancer-related mortality by 2025 due to its insidious onset, early distal metastasis, and dismal prognosis1,2. Metabolic disturbances of glucose, lipids, amino acids, etc., are significant contributors to this high mortality rate and major factors behind the disappointing outcomes of clinical trials targeting PDAC3,4. PDAC cells exhibit altered energy metabolism patterns, such as enhanced glycolysis, even in the presence of oxygen, a phenomenon known as the Warburg effect5. This metabolic shift supports the high energy demands of rapidly proliferating cancer cells, further interlinking with cancer hallmarks such as treatment resistance, metastasis, and invasion6. PDAC cells also extensively promote the metabolic rearrangement of non-cancer cells in the tumor microenvironment (TME) for nutrition support, clearance of metabolic waste, and immune escape so as to facilitate their growth and survival7,8. Efforts to target these metabolic aberrations in PDAC are being explored as potential therapeutic strategies.
Activated pancreatic stellate cell (PSC), the main stromal cell of PDAC, accounts for 50% of the stroma and is comprehensively implicated in PDAC metabolic imbalances, including reprogramming of stroma and energy metabolism9. The dense extracellular matrix (ECM) excessively secreted by PSC creates a physical hurdle that contributes to increased interstitial fluid pressure10, impaired hemoperfusion11, cellular hypoxia12, and blockage of nutrient and drug delivery13. Furthermore, metabolic interaction between PSC and PDAC cells is a crucial factor in PDAC progression14. PSC activated by PDAC cells rewrite its metabolic profile as aerobic glycolysis, producing nutrients such as lactate and non-essential amino acids to fuel the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) within PDAC cells, a phenomenon known as the reverse Warburg effect15. This metabolic symbiosis empahzsizes the intricate metabolic crosstalk and cooperation between stromal cells and cancerous cells within the TME, ultimately supporting PDAC growth and progression16.
Nuclear factor kappa B (NF-κB), a transcription factor, acts as a central integrator of various critical signaling pathways (e.g., PI3K/AKT and mTOR) and orchestrates the rearrangement of glucose, amino acid, lipid and mitochondrial metabolism in PDAC17. NF-κB responds very well to the glucose availability in cancer cells. Glucose deprivation can trigger the activation of NF-κB signaling, which upregulates the expression of glucose transporter 1 (GLUT1), thereby enhancing the glucose uptake of PDAC cells18. The activated NF-κB signaling then redirects cellular energy metabolism towards OXPHOS to optimize the utilization of available resources for the maintenance of cell survival and energy production, which is crucial for PDAC progression19. Therefore, the NF-κB signaling pathway and subsequent glucose metabolic adaptation could be a promising target for rectifying the abnormality of PDAC energy metabolism. Additionally, PDAC cells-derived galectin-3 has been reported to activate PSC and enhance ECM production through activation of the NF-κB signaling pathway, contributing to the crosstalk between the two cells and remodeling of the desmoplastic microenvironment20. The in-depth involvement of NF-κB signaling in metabolic reprogramming and matrix remodeling presents therapeutic opportunities to improve the treatment.
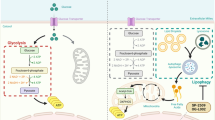
Here, we show a TME/PDAC cells sequential targeting nanosystem (T-AsiG-CPL) based on cationic liposome to co-deliver TPCA-1, a selective NF-κB inhibitor21, and a hybrid nucleotide sequence of CD71 aptamer and Glut1 siRNA (siG) linked by a disulfide bond (AsiG) to restore PDAC energy and stromal metabolism homeostasis. As shown in Fig. 1, T-AsiG-CPL disassembles in an acidic TME, leading to the release of TPCA-1 and AsiG. TPCA-1 induces the reversion of activated PSC to a quiescent state and decreases ECM deposition, facilitating secondary delivery of AsiG deep within the tumor tissue. AsiG subsequently targets PDAC cells that overexpress CD71 receptors and release siG in response to the high concentration of intracellular glutathione (GSH) upon disulfide bond breakage. Glut1 silencing reduces glucose uptake and mediates attenuation of aerobic glycolysis in PDAC cells, and the OXPHOS enhancement induced by NF-kB activation under glucose deprivation is further disrupted by TPCA-1. The combination of NF-kB suppression and Glut1 knockdown also synergistically interrupts the exchange of substance and energy between PSC and PDAC cells. The strategy, addressing abnormality of both stroma and energy metabolism, holds promise for improving treatment outcomes in challenging PDAC. In addition, it is a beneficial attempt to reveal the energy metabolism crosstalk between PSC and T3M4 cells, a human PDAC cell line characterized by overexpression of GLUT1, and modulate their crosstalk by tailor-designed nanosystem.

T-AsiG-CPL is formulated with cationic lipid (DOTAP), acidic cholesterol (CHEMS), and ionizable lipid (DSPE-PEOz), along with NF-κB inhibitor TPCA-1 and CD71 aptamer-modified siRNA targeting Glut1 (AsiG). TPCA-1 is liberated to the acidic tumor microenvironment (TME) by the pH-sensitive nanosystem (i), reversing the activated PSC to a quiescent state by blocking NF-κB pathway, which diminishes its metabolic support to PDAC cells and suppresses extracellular matrix (ECM) hyperplasia. CD71 aptamer subsequently mediates PDAC cell-targeted delivery of AsiG that is then cleaved by abundant GSH (ii) and releases Glut1 siRNA to disrupt the aerobic glycolysis inside the PDAC cells. Additionally, the inhibition of NF-κB by TPCA-1 curtails the compensatory increase in oxidative phosphorylation (OXPHOS) after glucose limitation in PDAC cells. This multifaceted nano-approach hinders the metabolic crosstalk between PSC and PDAC cells and potentiates the oncotherapy.
Results
Preparation and characterization of T-AsiG-CPL
Different pH-responsive components were tested to synthesize a desired cationic liposome for precise and rapid drug release in the weakly acidic (pH 6.5) TME. Cholesteryl hemisuccinate (CHEMS), an acidic cholesterol, can form a lipid bilayer with lecithin under neutral or alkaline conditions but is unstable in an acidic environment due to carboxylic acid protonation22,23. DSPE-PEOz (1, 2-distearoyl-sn-glycreo-3-phosphoetnanolmine-polyethylene oxide-zwitterion) is a pH-sensitive lipid with an ionizable zwitterionic group positively charged in low pH environments24. Liposomes based on CHEMS (CHEMS-Lipo), DSPE-PEOz (DSPE-PEOz-Lipo), or doped with both CHEMS and DSPE-PEOz (CHEMS/PEOz-Lipo, CPL) were obtained respectively by ethanol injection method (Supplementary Table 1). CPL was substantially disrupted within 30 min at pH 6.5. In contrast, it took at least 1 h for CHEMS-Lipo or DSPE-PEOz-Lipo to induce disassembly under the same conditions (Supplementary Fig. 1). Therefore, CPL was utilized as the pH-responsive liposome for subsequent experiments.
Transmission electron microscopy (TEM) imaging revealed that CPL had a spherical morphology with a particle size of 36.99 ± 4.89 nm (Fig. 2a), and its hydrodynamic diameter was 36.63 ± 3.23 nm with a positive surface charge of 9.53 ± 0.53 mV (Fig. 2b, c). Encapsulation of the hydrophobic small molecule drug TPCA-1 within the lipid bilayer of CPL resulted in TPCA-1-loaded CPL (T-CPL), which slightly increased surface charge of the nanoparticle to 15.93 ± 1.12 mV. The CPL/TPCA-1 mass ratio was further determined as 10:1 to obtain an optimal drug encapsulation efficiency of 56.53 ± 2.90% and a maximum drug loading efficiency of 5.14 ± 0.26% (Supplementary Table 2).

a Representative TEM images of the CPL-based nanoparticles from three independent experiments. Scale bar, 50 nm. b Size distribution of the CPL-based nanoparticles measured by dynamic light scattering. c Zeta potential of the CPL-based nanoparticles in PBS (pH 7.4) (n = 3 independent experiments). d Agarose gel electrophoresis assay of siRNA at various weight ratios of liposome to AsiG. e Quantitative analysis of the normalized siRNA intensities of AsiG-CPL and T-AsiG-CPL in the agarose gel electrophoresis assay from three independent experiments. f TEM images of CPL and T-AsiG-CPL at pH 6.5 were taken at different times in three independent experiments. Scale bar, 50 nm. g Cumulative release profiles of TPCA-1 from T-AsiG-CPL in PBS at pH 7.4 or 6.5 within 48 h (n = 3 independent experiments). h Agarose gel electrophoresis retardation assay of siG treated with 5 mM GSH for 2 h at pH 7.4 or 6.5 from three independent experiments. i siG protection assay of different nanosystems treated with RNase A for 2 h at pH 7.4 or 6.5 from three independent experiments. The data in (c, e, and g) are shown as the mean ± standard error (SEM). Source data are provided as a Source Data file.
Then, a CD71 aptamer (Apt) was covalently linked to a Glut1 siRNA (siG) via a disulfide bond to yield a DNA-siRNA hybrid sequence (AsiG) that imparted capabilities of PDAC cells-specific targeting (due to the overexpression of CD71 receptor on the PDAC cells) and GSH-responsive drug release to siG (Supplementary Table 3 and Supplementary Fig. 2). By electrostatic absorption with positively charged (2,3-Dioleoyloxy-propyl)-trimethylammonium-chloride (DOTAP), negatively charged AsiG was sequestered within the cationic liposome to form AsiG-CPL and T-AsiG-CPL. Agarose gel electrophoresis retardation assay demonstrated that over 90% retardation was achieved for both AsiG-CPL and T-AsiG-CPL at a ratio of 20:1 (CPL/AsiG, w/w), which was used for subsequent experiments (Fig. 2d, e). The hydrated particle size of T-AsiG-CPL increased to 59.6 ± 0.65 nm, and the surface charge was reversed to −12.2 ± 0.73 mV, demonstrating successful encapsulation of AsiG (Fig. 2b, c). Cryogenic electron microscopy (Cryo-EM) showed that T-AsiG-CPL possessed a spherical structure with a bilayer membrane (Supplementary Fig. 3 and Supplementary Table 4). Interestingly, AsiG loading promoted the TPCA-1 encapsulation efficiency of T-AsiG-CPL to 68.96 ± 1.62% by virtue of electrostatic interaction between negative AsiG and weakly basic TPCA-1 (Supplementary Table 2). T-AsiG-CPL exhibited good stability in PBS (10% serum) at 37 °C for 24 h, as evidenced by the unchanged spherical morphology (Supplementary Fig. 4).
Next, we investigated the acid-responsive behavior of T-AsiG-CPL by examining their morphological changes at pH 6.5 using TEM. Similar to CPL, T-AsiG-CPL lost the spherical shape and disassembled into fragments after 0.5 h incubation, demonstrating its rapid responsiveness in the TME (Fig. 2f). Both T-CPL and T-AsiG-CPL rapidly released TPCA-1 at pH 6.5, achieving an approximate cumulative release of 80% within 48 h (Fig. 2g and Supplementary Fig. 5). In contrast, less than 20% of TPCA-1 was detected at physiological pH 7.4, indicating restricted leakage in the blood circulation and normal tissues.
Then, the release of siG from T-AsiG-CPL was monitored by agarose gel electrophoresis after incubation with 5 mM GSH, a pathological concentration in PDAC cells. As shown in Fig. 2h, AsiG loaded in T-AsiG-CPL was completely retarded at pH 7.4, indicating good stability of cationic CPL consistent with the TEM results (Supplementary Fig. 4). Free siG could only be released when both weak acidity and GSH were fulfilled, which can be explained by the GSH-triggered breaking of the disulfide bond between siG and Apt after the liberation of AsiG resulting from the pH-responsive structural dissociation of T-AsiG-CPL (Fig. 2f). A sequentially responsive nanosystem is supposed to prevent the premature drug release during blood circulation and enable the controlled release of siG in PDAC cells. Furthermore, while naked AsiG was quickly degraded by RNase A, 90% of the CPL-encapsulated AsiG remained detectable, demonstrating the nanosystem’s effective protection against RNase A (Fig. 2i). All above results indicated successful preparation of T-AsiG-CPL, capable of stable delivery and programmed drug release by sequentially responding to TME and PDAC cells.
Targeted delivery of T-AsiG-CPL to PDAC cells
To explore the pH and CD71 aptamer dual-mediated cell-targeting of T-AsiG-CPL, we first screened various human PDAC cell lines using western blotting assay and selected T3M4 and BxPC-3 cells for subsequent experiments for their high expression of both CD71 and GLUT1 (Supplementary Fig. 6). The targeting potential of the nanosystem by PDAC cells was investigated using a Cy5-labeled siG (siG-Cy5) and visualized by confocal microscopy (Fig. 3a, b). After 1 h of co-incubation on ice at pH 6.5, most AsiG co-localized with the tumor cell membrane along with some red fluorescence dispersing in the cytoplasm, suggesting the aptamer-facilitated cell-targeting and cellular uptake of siG. Both AsiG-CPL and T-AsiG-CPL also showed fluorescence with comparable intensity and distribution to free AsiG, implying that AsiG was rapidly released from the pH-responsive liposome and targeted PDAC cells at pH 6.5. Pre-incubation of PDAC cells with CD71 antibody (anti-CD71) significantly decreased the cellular binding and uptake of AsiG, which, along with the results from flow cytometry (Fig. 3c–f), further confirmed the targeting of T-AsiG-CPL to PDAC cells mediated by the overexpressed CD71 aptamer. On the contrary, under the same conditions, only faint fluorescence was observed in PSC due to the undetectable expression of CD71 in these PDAC patient-derived cells (Supplementary Figs. 6, 7), which further ensures the cell specificity of siG to PDAC cells.

Confocal laser scanning microscopy images of T3M4 (a) and BxPC-3 (b) cells after AsiG-Cy5 (red)-loaded nanoparticles treatment at pH 6.5 for 1 h from three independent experiments (AsiG-Cy5: 250 nM). Nuclei were labeled with DAPI (blue), and cell membranes were labeled with Wheat Germ Agglutinin (WGA, green). anti-CD71, CD71 primary antibody. Scale bars, 20 μm. c Flow cytometry analysis of T3M4 cells after incubation with AsiG-Cy5 loaded formulations at pH 6.5 for 1 h (n = 3 independent experiments; AsiG-Cy5: 250 nM). d Mean fluorescence intensity (MFI) values of different formulations (n = 3 independent experiments). a. u., arbitrary units. e Flow cytometry analysis of BxPC-3 cells after incubation with AsiG-Cy5 loaded formulations at pH 6.5 for 1 h (n = 3 independent experiments, AsiG-Cy5: 250 nM). f MFI values of different formulations (n = 3 independent experiments). Cell viability of T3M4 cells (g), BxPC-3 cells (h), and PSC (i) following different treatments (AsiG: 50 nM; TPCA-1: 20 μM for human PDAC cells and 10 μM for PSC) at pH 7.4 or 6.5 for 48 h (n = 5 independent experiments). The data in (d) and (f–i) are displayed as the mean ± SEM. One-way ANOVA (d, f) and two-way ANOVA (g–i) with Bonferroni’s multiple comparisons test were used for statistical significance analysis. Source data are provided as a Source Data file.
Next, to validate the silencing efficiency of the Glut1 gene in PDAC cells, we screened three siG sequences and identified the most effective one for subsequent use (Supplementary Tables 3, 5 and Supplementary Fig. 8a, b). Following treatment with AsiG-CPL, an evident reduction in Glut1 mRNA and protein expression was observed in both T3M4 and BxPC-3 cells (Supplementary Fig. 8c–g). Remarkably, this reduction was comparable to the effect of commercially available lipofectamine 3000 (Lipo3000) transfection, underscoring the delivery capabilities of CPL and the potency of Glut1 silencing. Additionally, T-CPL inhibited GLUT1 expression, and the most pronounced silencing occurred when TPCA-1 was co-delivered with siG, proving the pivotal role of NF-κB in the regulation of GLUT125.
Furthermore, we evaluated the cytotoxicity of TPCA-1 in combination with Glut1 silencing against PDAC cells as well as PSC. CPL did not influence the viability of PDAC cells but increased the antiproliferative activity of TPCA-1 by virtue of promoting drug uptake (Fig. 3g, h). While both AsiG-CPL and AsiG-Lipo3000 presented moderate cytotoxicity (~20%), demonstrating the limited effect of single GLUT1 inhibition on PDAC cell viability. The T-AsiG-CPL group exhibited the highest inhibition (>60%) on the cell viability of PDAC cells, indicating a synergistic enhancement of siG to TPCA-1. Compared with the physiological pH environment, weak acidic culture conditions evoked a certain degree of reduction in cell activity in the T-AsiG-CPL group, emphasizing the sequentially responsive nanostrategy. Interestingly, the same treatments had no pronounced effect on PSC viability, which was consistent with the negligible cellular uptake of T-AsiG-CPL and verified the safety of TPCA-1 and siG for PSC (Fig. 3i and Supplementary Fig. 7). Collectively, T-AsiG-CPL exhibited selective cytotoxicity against the tumor cells without affecting the survival of surrounding PSC, thus avoiding the potential risk of PDAC metastasis mediated by simple depletion of PSC in the TME26.
PSC quiescence and ECM reduction in vitro
The metabolic crosstalk between PDAC cells and PSC leads to the overactivation of the NF-κB signaling pathway, which induces stroma remodeling and PDAC progression27. We verified the effects of the nanosystem on NF-κB in two PDAC cell lines, T3M4 and BxPC-3, as well as PSC cultured in conditional medium (CM) from each other at pH 6.5 to mimic the TME with close cell-cell interactions. Western blot and qPCR analysis demonstrated that phosphorylated NF-κB subunit p65 (p-p65) in both PDAC cells was significantly upregulated by the conditioned medium from PSC (PSC-CM) (p < 0.0001), while total p65 wasn’t obviously affected (Fig. 4a and Supplementary Fig. 9). Co-incubation with PSC-CM also increased nuclear p-p65 in the PDAC cells, indicating that overactivation of NF-κB is triggered by the PDAC cell-PSC crosstalk (Fig. 4b, c). A similar result was also observed in PSC treated with conditioned medium from T3M4 cells (T3M4-CM) (Fig. 4a, d, e). Free TPCA-1 and T-CPL allowed substantial inhibition of p-p65 expression in both PDAC cells and PSC. Notably, reduced glucose uptake caused by Glut1 silencing remarkably activated the negative feedback loop of NF-κB in PDAC cells28, confirmed by the upregulation of p-p65 by AsiG-CPL. Encouragingly, the co-delivery of TPCA-1 and siG by CPL suppressed this feedback enhancement on NF-κB in PDAC cells, establishing the foundation for subsequent energy metabolic regulation. In contrast, AsiG-CPL did not further activate the NF-κB signaling in PSC due to its lack of GLUT1 expression (Supplementary Fig. 6).

a Western blot for phosphorylated p65 (p-p65) and total p65 (p65) protein in whole cell lysates of T3M4 cells and PSC cultured with various treatments in standard culture medium or conditioned medium (CM) at pH 6.5 for 48 h. The normalized intensities of the p-p65 relative to p65 are presented. PSC-CM, CM from PSC; T3M4-CM, CM from T3M4 cells. b Immunofluorescence (IF) staining of p65 (green) in T3M4 cells after treatment with the indicated formulations for 48 h at pH 6.5. Nuclei were labeled with DAPI (blue). Scale bar, 10 μm. c Quantification of the normalized nucleus p65 protein expression in T3M4 cells (n = 6 independent experiments). d IF staining of p65 (red) in PSC treated with the indicated formulations for 48 h at pH 6.5. Nuclei were labeled with DAPI (blue), and cell skeletons were labeled with phalloidin (green). Scale bar, 25 μm. e Quantification of the normalized nucleus p65 protein expression in PSC (n = 6 independent experiments). f IF staining of α-SMA (green), fibronectin (green), and Sirius red staining (red) of deposited collagen of PSC treated with the indicated formulations for 48 h at pH 6.5. Nuclei were labeled with DAPI (blue). Scale bars, 25 μm. Quantification of the normalized fluorescence intensities of α-SMA (g), fibronectin (h), and collagen (i) in PSC. Cells were treated with various formulations (AsiG: 50 nM; TPCA-1: 20 μM for T3M4 cells and 10 μM for PSC, n = 3 independent experiments). Immunostaining experiments were independently repeated three times. The data in (c, e), and (g–i) are shown as the mean ± SEM. Two-way ANOVA with Bonferroni’s multiple comparisons test was used for statistical significance analysis. Source data are provided as a Source Data file.
Next, we assessed the capacity of T-AsiG-CPL for reverting activated PSC to a quiescent state. IF staining images provided compelling evidence that treatment with T-AsiG-CPL resulted in a striking reduction of α-SMA expression (Fig. 4f, g), a hallmark of activated PSC, which was in concordance with the observed NF-κB activation (Fig. 4d, e). Furthermore, following the induction of PSC quiescence, a dramatic decrease of fibronectin, a major ECM component, appeared upon T-AsiG-CPL treatment (Fig. 4f, h). Notably, the elevation of fibronectin expression induced by T3M4-CM was also substantially restrained. TPCA-1, T-CPL, or T-AsiG-CPL treatment significantly reduced collagen, another major ECM component secreted by PSC (Fig. 4f, i)
Considering the heterogeneity of PSC from different individuals, we validated NF-κB regulation and ECM inhibition by T-AsiG-CPL on two additional PDAC patient-derived PSC, namely PSC-2 and PSC-3. The results exhibited in these two cells were largely consistent with those observed in PSC-1 (Supplementary Figs. 10, 11), showing similar trends in the regulatory effects, though slight variations were noted. T-AsiG-CPL not only minimized the expression of phospho-p65, fibronectin, and collagen in PSC-2 and PSC-3 but also curbed the upregulation of these proteins triggered by indirect co-culture with PDAC cells, confirming the effective rectification of the nanosystem on the metabolism abnormality of PSC.
Modification of energy metabolic interaction between PSC and PDAC cells
Overactivation of GLUT1 and NF-κB helps mold the abnormal energy metabolism in PDAC together. We detected the efficacy of the nanostrategy in comprehensively modulating the energy metabolism disorder of PDAC cells. Compared to the control, Glut1 gene silencing together with NF-κB inhibition mediated by T-AsiG-CPL resulted in a ~40% decrease in glucose uptake and a ~ 100% reduction in lactate (a final metabolite of glycolysis) excretion of T3M4 cells (Fig. 5a, b). Consistently, co-delivery of TPCA-1 and siG by CPL induced the most remarkable reduction in both glycolysis (78.19%) and mitochondrial respiration (88.60%), which were measured by the extracellular acidification rates (ECAR) and oxygen consumption rates (OCR) respectively using a seahorse-based analysis (Fig. 5c, d and Supplementary Fig. 12a–e).

Glucose uptake (a) and lactate secretion (b) of T3M4 cells after different treatments in standard culture medium or PSC-CM at pH 6.5 for 48 h. Data were normalized to T3M4 cells cultured in the standard medium without treatment (n = 3 independent experiments). c Extracellular acidification rate (ECAR) of T3M4 cells treated with different formulations in standard culture medium (pH 6.5) for 48 h (n = 4 independent experiments). d Oxygen Consumption Rate (OCR) of T3M4 cells cultured in the standard medium (pH 6.5) after treatment of indicated formulations for 48 h (n = 6 independent experiments). e ECAR of T3M4 cells treated with different formulations in PSC-CM for 48 h at pH 6.5 (n = 6 independent experiments). f OCR measurements of T3M4 cells cultured in PSC-CM (pH 6.5) for 48 h (n = 6 independent experiments). g ECAR of T3M4 cells treated with T-AsiG-CPL in standard medium or PSC-CM for 48 h at pH 6.5 (n = 12 independent experiments). h Glycolysis and glycolytic capacity of T3M4 cells were calculated from the ECAR curves (n = 12 independent experiments). i OCR measurements of T3M4 cells treated with T-AsiG-CPL cultured in standard medium or PSC-CM (pH 6.5) for 48 h (n = 12 independent experiments). j Basal respiration, maximal respiration, and ATP production of T3M4 cells were calculated from the OCR curves (n = 12 independent experiments). Cells were treated with various formulations (AsiG: 50 nM; TPCA-1: 20 μM). The data are shown as the mean ± SEM. Two-way ANOVA (a, b), (h), and (j) with Bonferroni’s multiple comparisons test were used for statistical significance analysis. Source data are provided as a Source Data file.
Although PDAC cells primarily depend on glycolysis for 85% of their energy production5, the substantial presence of PSC in the TME drove a reprogramming in the energy metabolism of T3M4 cells from glycolysis to OXPHOS (Fig. 5e, f and Supplementary Fig. 12f–j). Co-incubation with PSC-CM at pH 6.5 induced a decrease in glycolysis activity from 59.42 to 26.33 mpH/min (Fig. 5g, h) and an increase of basal respiration from 66.71 to 96.64 pMol/min (Fig. 5i, j) in T3M4 cells, accompanied by reductions in both glucose uptake (Fig. 5a) and lactate secretion (Fig. 5b). This switch of energy patterns indicates an extra metabolic support of activated PSC to T3M4 cells growth. However, it was significantly curbed by the designed dual inhibition of NF-κB to restore quiescence in PSC and block OXPHOS enhancement in T3M4 cells. Similar metabolic shifts were observed in the crosstalk between BxPC-3 cells and PSC, where T-AsiG-CPL also demonstrated significant dual metabolic suppression (Supplementary Fig. 13).
Considering the pivotal role of PSC in reprogramming the energy metabolism of PDAC cells, we further analyzed the regulatory effects of T-AsiG-CPL on the bioenergetics profile of PSC. Specifically, metabolic plasticity was observed in PSC following treatment with T-AsiG-CPL, characterized by a moderate inhibition of glucose uptake (Fig. 6a), lactate secretion (Fig. 6b), and ECAR (Fig. 6c and Supplementary Fig. 14a, b), as well as a significantly reduced OCR (Fig. 6d and Supplementary Fig. 14c–e). Notably, AsiG-CPL did not contribute to altering the glycolysis flux of PSC, while T-CPL reduced both glycolysis and basal OCR by about 30%. On account of the undetectable expression of GLUT1 in PSC (Supplementary Fig. 6), we hypothesized that the dual inhibitory effect in glycolysis and OXPHOS stemmed predominantly from PSC quiescence induced by NF-κB inhibition (Fig. 4a, d, e). Importantly, T-AsiG-CPL did not disturb the viability of PSC (Fig. 3i), reinforcing the correlation between the decrease in energy metabolism and the inhibition of PSC activation. Furthermore, contrary to the effect of PSC-CM on PDAC cells, T3M4-CM prompted a reprogramming of PSC from OXPHOS to glycolysis (Fig. 6e–j and Supplementary Fig. 14f–j) and enhanced lactate secretion (p < 0.001, Fig. 6b), a manifestation of T3M4-induced activation of PSC15. Encouragingly, T-AsiG-CPL treatment notably inhibited this metabolic shift and restored PSC energy metabolism upregulated by PDAC cells (Fig. 6g-j).

Glucose uptake (a) and lactate secretion (b) of PSC after different treatments in standard culture medium or T3M4-CM at pH 6.5 for 48 h. Data were normalized to PSC cultured in the standard medium without treatment (n = 3 independent experiments). c ECAR of PSC treated with different formulations in standard culture medium (pH 6.5) for 48 h (n = 6 independent experiments). d OCR of PSC cultured in the standard medium (pH 6.5) after treatment of indicated formulations for 48 h (n = 6 independent experiments). e ECAR of PSC treated with different formulations in the T3M4-CM (pH 6.5) for 48 h (n = 6 independent experiments). f OCR of PSC cultured in the T3M4-CM (pH 6.5) after treatment of indicated formulations for 48 h (n = 6 independent experiments). g ECAR of PSC treated with T-AsiG-CPL in standard culture medium or T3M4-CM for 48 h at pH 6.5 (n = 6 independent experiments). h Glycolysis and glycolytic capacity of PSC were calculated from the ECAR curves (n = 6 independent experiments). i OCR measurements of PSC with T-AsiG-CPL treatment cultured in standard culture or T3M4-CM (pH 6.5) for 48 h (n = 6 independent experiments). j Basal respiration, maximal respiration, and ATP production of PSC were calculated from the OCR curves (n = 6 independent experiments). Cells were treated with various formulations (AsiG: 50 nM; TPCA-1: 10 μM). The data are shown as the mean ± SEM. Two-way ANOVA (a, b), (h), and (j) with Bonferroni’s multiple comparisons test were used for statistical significance analysis. Source data are provided as a Source Data file.
These data suggest that dual inhibition of GLUT1 and NF-κB in both PDAC cells and activated PSC by T-AsiG-CPL effectively curtails two crucial energy pathways, glycolysis, and OXPHOS, and blocked energy rewriting induced by the intercellular crosstalk so as to cope with the rapid growth of PDAC cells (Fig. 3g, h).
Blood pharmacokinetics and biodistribution of T-AsiG-CPL in vivo
Encouraged by the effective PDAC cell growth inhibition and PSC quiescence in vitro, we further investigated the pharmacokinetics and bio-distribution of T-AsiG-CPL in an orthotopic pancreatic tumor mouse model, which was established by co-inoculation of mice with T3M4-luci cells and PSC that has been demonstrated to closely resemble human pancreatic cancer by generating desmoplastic stroma29,30. The circulation time of T-AsiG-CPL in the mice was determined by measuring the fluorescence intensity of Cy5 in blood after intravenous injection of siG-Cy5, AsiG-Cy5, and T-AsiG-Cy5-CPL, respectively (Fig. 7a, b). Using a two-compartment model, the elimination half-lives (t1/2, elim) of free siG-Cy5, AsiG-Cy5, and T-AsiG-Cy5-CPL were calculated to be 1.35, 1.74 and 7.83 h by DAS 2.0, respectively (Supplementary Table 6). Compared to the rapid blood elimination of free siG-Cy5 and AsiG-Cy5, the nanosystem greatly prolonged the circulation time of siG-Cy5 in T-AsiG-Cy5-CPL, indicating that CPL can effectively protect and deliver siG to the tumor site during circulation.

a Fluorescence images of blood samples from the tumor-bearing mice treated with siG-Cy5, AsiG-Cy5, or T-AsiG-Cy5-CPL. b Pharmacokinetic curves of siG-Cy5, AsiG-Cy5, and T-AsiG-Cy5-CPL in the blood, shown as the percentage of the injected dose (% ID) (n = 3 mice). Ex vivo fluorescence images of tumors and major organs at 24 h (c) and 48 h (e) following i.v. administration of siG-Cy5, AsiG-Cy5 or T-AsiG-Cy5-CPL. The average fluorescence intensities in tumors and major organs at 24 h (d) and 48 h (f), (n = 3 mice). The mice were administered various fluorescent formulations (siG: 1.0 mg/kg; TPCA-1: 20 mg/kg) via tail vein injection. The data in (b, d, f) are presented as the mean ± SEM. Two-way ANOVA (d, f) with Bonferroni’s multiple comparisons test was used for statistical significance analysis. Source data are provided as a Source Data file.
The biodistribution of siG-Cy5, AsiG-Cy5, and T-AsiG-Cy5-CPL in vivo was then evaluated through ex vivo fluorescence imaging of the tumors and major organs at 24 and 48 h post-administration. T-AsiG-Cy5-CPL exhibited 2.81-fold and 2.62-fold stronger fluorescent signal in tumors after 24 h compared to siG-Cy5 (p < 0.001) and AsiG-Cy5 (p < 0.01), respectively, indicating improved tumor accumulation of siG by the nanocarrier (Fig. 7c, d). After 48 h of T-AsiG-Cy5-CPL injection, a 6.85% enhancement in fluorescence was observed in tumors in comparison with that at 24 h, suggesting a sustained drug concentration within 48 h by virtue of a much higher expression of CD71 in the tumors than in other major organs (Fig. 7e, f, Supplementary Fig. 15). Notable fluorescence detected in the livers after T-AsiG-Cy5-CPL treatment may be attributed to the preference of liver Kupffer cells and liver sinusoidal endothelial cells for liposome31,32. Moreover, strong signals were observed in the kidneys after treatments of siG-Cy5 and AsiG-Cy5, which might mean the renal clearance of siG-Cy5 from the disassembled CPL33. Collectively, the remarkably intratumoral accumulation of T-AsiG-CPL demonstrated the potential application of the programmed targeting nanosystem in the desmoplastic PDAC TME.
Stromal modulation in desmoplastic PDAC
The stromal modulation efficacy of T-AsiG-CPL in vivo was evaluated by treating the T3M4/PSC tumor-bearing mice with different formulations intravenously every two days for six injections (Fig. 8a). Remarkably, immunohistochemical (IHC) staining revealed that the nano-delivery reduced the α-SMA-positive area from 67.16% (TPCA-1) to 42.88% (T-CPL), demonstrated the effective delivery of pH-sensitive CPL within the TME (Fig. 8b, c). Nevertheless, AsiG-CPL did not trigger a statistically significant difference in α-SMA expression, which is in accordance with the observations made in PSC in vitro (Fig. 4f, g). T-AsiG-CPL also caused a significant reduction of α-SMA levels (p < 0.0001). At the same time, the maximum decrease in ECM abundance was also exhibited in the tumors receiving T-AsiG-CPL treatment, which was confirmed by a minimal distribution of fibronectin and collagen, indicating the effective modulation to stromal metabolism facilitated by PSC quiescence (Fig. 8b, d, e).

a Schematic illustration of the treatment procedures and the grouping description. T3M4-luci/PSC orthotopic xenografted mice in different groups were treated with Saline (G1), CPL (G2), TPCA-1 (G3), T-CPL (G4), AsiG-CPL (G5), TPCA-1 + AsiG-CPL (G6), or T-AsiG-CPL (G7) (siG: 1.0 mg/kg; TPCA-1: 20 mg/kg). b Representative histological analysis with immunohistochemical (IHC) staining of α-SMA (brown), fibronectin (brown), and two-photon autofluorescence of collagen (green) in tumor sections. Scale bars, 50 μm. Quantitative analysis of the normalized α-SMA (c), fibronectin (d), and collagen (e) protein expression in tumors after different treatments (n = 3 mice). A total of 6 images for each group (two random fields for each tumor section, three mice per group) were analyzed semi-quantitatively. Schematic illustration (f) and three-dimensional reconstruction (g) of orthotopic pancreatic tumors at the starting and endpoint of the T-AsiG-CPL treatment, imaged with a two-photon microscope at 1150 nm excitation wavelength after intravenously injecting siG-Cy5 or T-AsiG-Cy5-CPL (1.0 mg/kg siG and 20 mg/kg TPCA-1) into the mice for 24 h, (n = 3 mice). Scale bar, 100 μm. h The corresponding intensity profile of the Cy5-labeled siG at the tumor depth of 0–120 μm (n = 3 mice). The data in (c-e) and (h) are presented as the mean ± SEM. One-way ANOVA with Bonferroni’s multiple comparisons test was used for statistical significance analysis. Source data are provided as a Source Data file.
To verify the potential of the nanosystem-enhanced drug delivery after stromal modulation, we visualized the penetration behavior of siG-Cy5 in the orthotopic T3M4-luci/PSC tumors with or without T-AsiG-CPL treatment. As shown in Fig. 8f, g, three-dimensional reconstruction of images revealed very sparse signals of siG-Cy5 fluorescence in tumors without T-AsiG-CPL treatment. The nanosystem only enabled the enhancement of siG-Cy5 accumulation in the superficial layer (0-40 μm) of the untreated tumors due to its limited access to the deep tumor (Fig. 8h). After 6 treatments with T-AsiG-CPL (Fig. 8a), the tumor penetration of siG-Cy5 was remarkably improved by virtue of the decrease of ECM (Fig. 8g, h). Nevertheless, abundant fluorescence of T-AsiG-CPL was monitored at a maximum depth of 120 μm in tumors at the endpoint of the treatment, filling over 56.10% of the tumor volume (Fig. 8g, h and Supplementary Fig. 16), implying the potential of the nanosystem for improving in vivo drug delivery through the sequential tumor targeting strategy.
Anticancer effect of T-AsiG-CPL in vivo
Benefiting from the remarkable potential of T-AsiG-CPL to enhance drug penetration and localized accumulation within the tumor by reducing ECM abundance, we investigated its antitumor efficacy mediated through the dual inhibition of energy metabolism. An orthotopic PDAC mouse model was first established by co-inoculating luciferase-expressing T3M4 cells and PSC, which enabled us to monitor the tumor progression using in vivo bioluminescence imaging during treatment (Fig. 9a, b). The weight of the tumors treated with T-CPL was reduced by 66.72% compared to the saline group, which was approximately 2-fold higher than that of TPCA-1 (26.33%) or TPCA-1 plus AsiG-CPL (33.59%), thus demonstrating the efficacy of NF-κB inhibition enhanced by the effective delivery (Fig. 9c–e). AsiG-CPL treatment strongly restrained the tumor growth, which derived from a 50% decrease in GLUT1 expression within the tumor (Fig. 9f, g). The smallest tumor resulted from T-AsiG-CPL treatment, with a 79.13% reduction in tumor weight and an 82.79% loss of tumor volume (Fig. 9c–e). Consistent with this, according to the histological staining of the tumor sections, a loose cell arrangement with the lowest expression of GLUT1, p65, and Ki67 was observed in the T-AsiG-CPL group, confirming the dual inhibition of ECM and tumor cell energy metabolism (Fig. 9f–i). Furthermore, we detected the tumor metastasis on mesenteries, livers, and lungs. As shown in Supplementary Fig. 17, no metastatic foci were observed after treatment with either TPCA-1 plus AsiG-CPL or T-AsiG-CPL, suggesting that PSC quiescence-based stromal modulation does not elevate the risk of T3M4 orthotopic tumor metastasis. No significant difference was observed in body weight between the control and any experimental groups (Supplementary Fig. 18a). Meanwhile, none of the formulations disturbed the liver and kidney functions of the mice according to serum biochemistry measurements (Supplementary Fig. 18b). More importantly, no abnormalities of histomorphology were induced in major organ tissue after various treatments (Supplementary Fig. 19), suggesting good biocompatibility and biosafety of the nanosystem. Collectively, these data provide strong evidence for the potential of T-AsiG-CPL in enhancing PDAC treatment through dual suppression of energy metabolism and PSC re-education.

a Representative bioluminescence images of the T3M4-luci/PSC orthotopic xenografted mice on days 6, 12, and 18. The mice in different groups were treated with Saline (G1), CPL (G2), TPCA-1 (G3), T-CPL (G4), AsiG-CPL (G5), TPCA-1 + AsiG-CPL (G6), or T-AsiG-CPL (G7) (siG: 1.0 mg/kg; TPCA-1: 20 mg/kg). Images were randomly selected from 6 mice in each group. b Quantitative analysis of the luminescence signal in vivo (n = 6 mice). c Image of the excised orthotopic tumors with spleens. Tumor weight (d) and volume (e) at the endpoint of the treatments (n = 6 mice). f Histological studies of tumor sections with IHC staining of GLUT1 and p65, H&E staining, and IHC staining of Ki67. Scale bars, 50 μm. Quantification of GLUT1 (g), p65 (h), and Ki67 (i) expression in tumors (n = 3 mice). A total of 6 images for each group (two random fields for each tumor section, three mice per group) were analyzed semi-quantitatively. The data are displayed as the mean ± SEM. One-way ANOVA with Bonferroni’s multiple comparisons test was used for statistical significance analysis. Source data are provided as a Source Data file.
Discussion
PDAC is an exceptionally aggressive tumor that progresses through the interplay of cancerous and non-cancerous cells within the TME2,34. The failure of current clinical applications that target only tumor cells and ignore TME also illustrates that PDAC is not an island of tumor cells35. PSC, which constitutes 50% of PDAC stroma, is a pivotal contributor to this microenvironment and tumor malignancy35,36. The abundant ECM from PSC promotes a state of nutrient deprivation that forms a distinct mechanical barrier to drug delivery by squeezing the blood vessels and reducing hemoperfusion3,37. Intercellular metabolic crosstalk in TME encourages PSC to undergo a reverse Warburg effect driven by PDAC cells15. Then, PSC secretes nutrients to feed PDAC cells, prompting the energy conversion of the PDAC cells from glycolysis towards OXPHOS, thus supporting PDAC progression7. Although this metabolic rewiring for environmental adaptation has triggered a burgeoning development of metabolic inhibition strategies, more personalized therapeutic strategies still need to be explored in PDAC treatment.
Studies have demonstrated heterogeneity in PDAC metabolism, which implies differences in tumor metabolic preferences among different patient sources or at different spatial and temporal states of the same patient14. Based on the analysis of energy metabolic profiles, PDAC cell lines have been categorized into three groups so far: glycolytic group, OXPHOS group, and less metabolically active group38. Metabolic heterogeneity necessitates personalized treatment for PDAC. Furthermore, glucose serves as the primary nutrient donor for PDAC metabolism and biosynthesis, with key metabolic enzymes controlling these pathways4. Although these enzymes have been considered attractive therapeutic targets, single inhibition on them is insufficient to completely suppress PDAC progression39,40. This is attributed to the flexible compensatory mechanism adopted by PDAC to overcome metabolic perturbations, which includes increasing expression of alternative enzyme isoforms and dependencies41,42 on alternative metabolic pathways, such as glutamine metabolism43,44,45. These insights provide a basis for designing more effective therapeutic regimens. The modifiability and functional integration of customized nanomaterials present a promising opportunity to simultaneously block multiple metabolic pathways, and a variety of strategies have been developed to modulate tumor metabolism (glycolysis or fatty acid synthesis pathway) in combination with chemotherapy, radiotherapy, or immunotherapy to achieve multiple-punch to tumors46,47. Despite the current focus of most nanomedicines on regulating PDAC cells individually, there remains a largely untapped potential in harnessing the functional integration advantages of nanodrugs for multicellular or intercellular metabolic modulation45,48.
In the current study, we delineate the design and functional characterization of a pH/GSH-sensitive cationic liposome formulation, T-AsiG-CPL, which enables effective co-delivery of TPCA-1 and a PDAC cell-targeting Glut1 siRNA, allowing targeted recalibration of abnormal energy and stromal metabolism in PDAC. It was observed that T-AsiG-CPL rapidly responds to the TME, thereby facilitating non-selective uptake of TPCA-1 by both PDAC cells and PSC, leading to an effective attenuation of NF-κB signaling (Fig. 4). Consequent to the inhibition of the NF-κB pathway, both α-SMA expression of PSC and ECM deposition were downregulated. This modulation effectively restored stromal homeostasis, as evidenced by the enhanced drug permeability in a desmoplastic PDAC xenograft tumor model, detailed in Fig. 8f-h. Previous studies have demonstrated that TPCA-1 helps maintain the normal phenotype of fibroblasts and prevents their fibrotic transformation49,50. Other drugs, such as all-trans retinoic acid (ATRA), have also been reported to restore activated PSC to a quiescent state and increase chemosensitivity in PDAC51. However, we ultimately chose TPCA-1 not only for its ability to induce PSC quiescence but also because it plays a crucial role in PDAC cells by inhibiting the compensatory activation of NF-κB. Under low glucose conditions, glutamate dehydrogenase 1 (GDH1) phosphorylats and interacts with p65 and IKKβ of the NF-κB signaling pathway, and α-ketoglutarate produced by GDH1 binds to and activates IKKβ and NF-κB signaling, leading to compensatory glucose uptake by upregulating GLUT118. This stress response, aimed at regulating cell survival and responding to environmental pressures, was adequately revealed in PDAC cells in this study (Fig. 4a–c and Supplementary Fig. 9a). Nonetheless, Glut1 knockdown did not lead to activation of NF-κB signaling in PSC due to its undetectable GLUT1 (Fig. 4d–e and Supplementary Fig. 6). On the contrary, AsiG was preferentially taken in by PDAC cells overexpressing CD71 receptor (Supplementary Fig. 6). The diverse cytotoxicity of T-AsiG-CPL to PDAC cells and PSC further underlines good biosafety and accuracy of the strategy (Fig. 3g–i), and minimizes the potential risk of PDAC metastasis that might arise from simple PSC depletion26.
GLUT1 suppression leads to reduced glucose uptake and lactate secretion, thereby disrupting the overwhelming glycolytic flux in PDAC cells (Fig. 5a–c and Supplementary Fig. 12a, b). Notably, the classic negative feedback loop characterized by NF-κB-mediated upregulation of GLUT1 in response to glucose deprivation was also fully obstructed by the potent Glut1 silencing resulting from the well-designed multiple inhibition nanostrategy. NF-κB is a cellular energy sensor, orchestrating the balance between glycolysis and OXPHOS to ensure metabolic energy homeostasis and adaptation to environmental changes52. In this context, Birkenmeier et al. have shown that NF-κB can facilitate a metabolic shift toward OXPHOS in Hodgkin lymphoma cells25. In stark contrast, TPCA-1 disrupts both glycolytic and OXPHOS in PDAC cells in this study (Fig. 5c, d and Supplementary Fig. 12). We hypothesize that the diversity is probably derived from the intricate interaction between the NF-κB signaling pathway and p53 in various cells53. This is supported by the work of Johnson et al., which demonstrated that NF-κB suppression in p53-positive cancer cells diminished oxygen consumption and ATP synthesis. Conversely, in p53-negative cancer cells, NF-κB knockdown was associated with an upregulation of OXPHOS and ATP levels. While the role of NF-κB in metabolic regulation is complex, there is no doubt that the activation of NF-κB can rewire cellular metabolic networks to maintain and promote cell survival.
The abundance of PSC within the TME facilitates cellular crosstalk that promotes energy reprogramming in PDAC cells, where the energy metabolism of PDAC cells shifts from glycolysis to OXPHOS (Fig. 5g–j). This metabolic reprogramming of PDAC cells is regulated by NF-κB overactivation induced by co-culture with PSC. Therefore, the combinational suppression of both GLUT1 and NF-κB by T-AsiG-CPL realized the dual intervention of PDAC cells and PSC and co-blocking of major energy metabolic pathways (glycolysis and OXPHOS) in PDAC (Fig. 5). Considering the negligible expression of GLUT1 and low uptake of AsiG in PSC, we speculate that the change in PSC energy metabolism is largely due to TPCA-1-induced quiescence.
While encouraging results are demonstrated in this study, there are still some limitations. First, different PSC populations in the same pancreatic TME exhibit phenotypic diversity and functional heterogeneity, which play essential roles in PDAC progression and therapy response54,55,56. It is crucial to modulate the unique functions of these PSC populations in therapies targeting the metabolic crosstalk between the PSC and PDAC cells, but not yet been considered in this study. Second, despite effective treatment conducted here by simultaneously inhibiting glucose metabolism and the compensatory OXPHOS, metabolic therapy remains challenging in PDAC due to the complicated nutrient sources and energy supplies45. A comprehensive understanding of the interactions among different metabolic pathways and their roles in PDAC progression is essential for developing strategies that can precisely target tumor metabolism. Finally, although PDAC research including the current study is increasingly directed toward personalized treatments, several challenges, such as genetic variability among individuals, the complexity of the TME, and variability in treatment responses, persist. Additionally, cost-effectiveness, clinical feasibility, and the durability of long-term effects are also critical issues that future research needs to address.
In summary, we introduce a programmed targeting nanosystem based on a sequentially responsive liposome to dual rectify the abnormal energy and stromal microenvironments via coordinated regulation of GLUT1 and NF-κB signal in PDAC cells and PSC, thus improving the anti-cancer efficacy of PDAC. The delivery of TPCA-1 by the TME-targeting nanosystem effectively converts activated PSC to a quiescent state, disrupting its metabolic support to PDAC cells and reducing ECM hyperplasia. This facilitates deeper penetration of the PDAC cell-targeting siRNA into the tumor, which then inhibits GLUT1 overexpression and disrupts aerobic glycolysis of PDAC cells. Concurrently, the OXPHOS enhancement triggered by NF-κB overactivation under glucose scarcity is further hampered by TPCA-1. Our detailed analysis of the interaction between PSC and PDAC cells underscores the effectiveness of this nanosystem in interrupting intercellular energy and substance exchange. The tailor-designed modulation of metabolic crosstalk leads to effective suppression of tumor progression in an orthotopic xenograft mouse model of PDAC. This points towards a more personalized approach for PDAC treatment, considering the diverse energy metabolic profiles of the tumor.
Methods
Our research complies with all relevant ethical regulations. All animal experiments are approved by the Institutional Animal Care and Use Committee of the National Center for Nanoscience and Technology, China.
Materials
Hydrogenated soy phosphatidylcholine (HSPC, N01003) and (2,3-Dioleoyloxy-propyl)-trimethylammonium-chloride (DOTAP, O02002) were purchased from A.V.T. Pharmaceutical Tech Co., Ltd. (Shanghai, China). Cholesteryl hemisuccinate (CHEMS, 361932) was from Shanghai Yuanye Bio-Technology Co., Ltd. (Shanghai, China). Cholesterol (156754) was obtained from J&K Scientific Co., Ltd. (Guangzhou, China). DSPE-mPEG2000 (LP-R4-039), and DSPE-PEOz2000 (R-PL2056-2) were acquired from Ruixibio Co., Ltd. (Xian, China). Dulbecco’s Modified of Eagle Medium/F12, (DMEM/F12, 219-000), RPMI 1640 Medium (350-030), fetal bovine serum (FBS, 086-150), penicillin, and streptomycin were from Wisent Biotechnology Co., Ltd. (Montreal, Canada). 2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide (TPCA-1, HY-10074) was purchased from MedChem Express LLC. (Shanghai, China). Oligomycin (ab141829), antimycin A (ab141904), and iFluor 488-labeled phalloidin (ab176753) were obtained from Abcam (Cambridge, UK). Glutathione (GSH, PHR1359), glucose (G7528), glutamine (G8540), sodium pyruvate (S8636), 2-Deoxy-D-glucose (2-DG, D8375), trifluoromethoxy carbonyl cyanide phenylhydrazone (FCCP, C2920), and rotenone (R8875) were from Sigma-Aldrich (USA). D-Luciferin potassium salt (D812647) was from Macklin Inc. (Shanghai, China). Dimethyl sulfoxide (DMSO, D8371), DNase/RNase-free water (R1600), RNase A (R8020), Phosphate Buffered Saline (PBS, P1020), Dulbecco’s phosphate Buffered Saline (D-PBS, D1040), Tween-80 (T8360), sodium dodecyl sulfate (SDS, S8010), Radio Immunoprecipitation Assay (RIPA, R0010), Phenylmethylsulfonyl fluoride (PMSF, P0100), yeast-tRNA (9014-25-9), and 4’,6-diamidino-2-phenylindole (DAPI, C0065) were obtained from Solarbio Life Science (Beijing, China). 4SGelRed (A616697), and CD71 Aptamer (Apt) were acquired from Sangon Biotech Co., Ltd. (Shanghai, China). Glut1 siRNA (siG), Negative Control siRNA (siN.C), CD71 Aptamer-SS-Glut1 siRNA (AsiG), and siG-Cy5 were customized from GenePharma Technology Co., Ltd. (Suzhou, China). Reduced serum medium (Opti-MEM, 31985), and Lipofectamine 3000 Transfection Reagent (L3000-015) were obtained from Invitrogen of Thermo Fisher Scientific Inc. (Waltham, USA). Matrigel (354234) was obtained from Corning (New York, USA). 6×Loading Buffer (9156), and 20 bp DNA Ladder (3420 A) were purchased from TaKaRa Biomedical Technology Co., Ltd. (Beijing, China). iFluor® 488-Wheat Germ Agglutinin (WGA) conjugate (25530) was from AAT Bioquset Inc. (USA). Sterile syringe filters, 0.45 μm (slhv033rs), and ultrafiltration centrifuge tube (MW = 100 KD, UFC801096) were obtained from Millipore (Germany). Dialysis bags (= 10 KD, HF131417) were from Spectrum (California, USA). Rabbit polyclonal primary antibodies against NF-κB p65 (10745-1-AP), glucose transporter 1 (GLUT1, 21829-1-AP), smooth muscle actin (α-SMA, 14395-1-AP), fibronectin (15613-1-AP) were purchased from Proteintech Group, Inc. (Chicago, USA). Mouse monoclonal primary antibody against NF-κB p65 (sc-8008) and Ki67 (sc-23900) was from Santa Cruz Biotechnology (Dallas, USA). Rabbit polyclonal primary antibody p-p65 (Ser536, 3033 T), Alexa Fluor® 488 (4412S) or Alexa Fluor® 594 (8889) -conjugated goat anti-rabbit IgG (H + L), F(ab’)2 Fragment were from Cell Signaling Technology (Danvers, USA).
Preparation of T-AsiG-CPL
Liposomes were prepared using the ethanol injection method as described by Pons et al.57. In brief, HSPC, DOTAP, CHEMS, and DSPE-PEOz were fully dissolved in ethanol at a molar ratio of 20:40:35:5 to obtain a 50 mg/mL mixture. The mixture was then combined with TPCA-1 at a mass ratio of 10:1 and slowly injected into PBS containing AsiG while being sonicated. The milky emulsion was obtained through continuous sonication for 20 min. T-AsiG-CPL was acquired by removing ethanol and unencapsulated TPCA-1 and AsiG via centrifugation (2000 g × 20 min, 4 °C) in ultrafiltration tubes (Mw = 100 KD), followed by resuspension in PBS.
TPCA-1 loading, encapsulation, and release
TPCA-1 was dissolved in DMSO to create a 100 mg/mL master mix. TPCA-1-loaded T-CPL and T-AsiG-CPL were prepared by incorporating various amounts of TPCA-1 into the ethanol mixture of the lipid compositions according to the above method. Ethanol and free drug were eliminated through centrifugation (2000 g × 20 min, 4 °C) using ultrafiltration tubes (Mw = 100 KD), followed by resuspension in 0.5 mL PBS, lysis of liposome with 0.5 mL ethanol and filtration through a microporous membrane with a pore size of 0.22 μM to determine the concentration of TPCA-1 via high-performance liquid chromatography (detection wavelength: 310 nm, mobile phase: 30% acetonitrile/70% water). The equation was calculated as follows:
TPCA-1 released from T-CPL and T-AsiG-CPL were collected in PBS at pH 7.4 or 6.5, respectively. Specifically, 1 mL of liposome was dialyzed against a solution of 30 mL PBS containing 1% Tween-80 using a dialysis bag (Mw = 10 KD) with an oscillation speed of 100 rpm at 37 °C. The outside buffer was collected over time and replenished with fresh buffer of the same volume. TPCA-1 concentration of collected samples was quantified by High Performance Liquid Chromatography, with detection at 310 nm (water/acetonitrile: 20%/80%, v/v, flow rate: 1 mL/min).
siRNA loading, release, and protection
For the siRNA adsorption assay, AsiG-CPL or T-AsiG-CPL was synthesized by mixing AsiG with lipid mixture at various w/w ratios with sonication for 20 min. The resulting nanoparticles were mixed with 6 × Loading buffer and analyzed by 1.5% agarose gel electrophoresis and then analyzed by a ChemiDoc imaging analyzer (Bio-Rad, USA). For the detection of siG release, AsiG-CPL or T-AsiG-CPL were incubated with 5 mM GSH at pH 7.4 or 6.5 for 2 h. In the siRNA protection experiment, T-AsiG-CPL was co-incubated with RNase A (0.1 mg/mL) at 37 °C for 2 h before being treated with 1% SDS for an additional 10 min. The resulting samples were visualized after agarose gel electrophoresis.
Characterization of nanoparticles
The morphology of nanoparticles was observed via transmission electron microscopy (TEM, Ht-7700, Hitachi). Samples were prepared by dropping 10 μL suspended nanoparticles (0.2 mg/mL) onto carbon-coated copper grids with a deposit for 10 min, followed by negative staining with 2% uranyl acetate for 20 min. The morphology of the liposomes was also characterized by a cryo-electron microscope (FEI Talos F200C, Thermo Fisher). A 3–5 µL liposome suspension (20 mg/mL) was added to a hydrophilically treated copper grid for 20 s, followed by blotting of the excess liquid, and was immediately plunge-frozen in liquid ethane. The grid was then stored in liquid nitrogen until imaging. Samples were imaged using a FEI Talos Arctica microscope at 200 kV. Images were captured using a bottom-mount K3 camera (Gatan, Inc., Pleasanton, CA) at 36,000 × magnification (0.5705 Å/pixel). Image analysis was performed using the CryoSPARC v.4.5.3 software. The zeta potential and hydrodynamic diameter were determined using dynamic light scattering (DLS, Zetasizer, Malvern).
Cell culture
The human PDAC cell lines T3M4, Panc-1, BxPC-3, and Aspc-1 cells were acquired from the American Type Culture Collection (ATCC). T3M4 and Panc-1 cells were authenticated by the short tandem repeat fingerprinting (STR). Luciferase-expressing T3M4 (T3M4-luci) cells were constructed using lentivirus (GeneChem, China) transduction and screened with puromycin (2 μg/mL). All cells were cultured in RPMI 1640 Medium with 10% FBS, and 1% penicillin/streptomycin in a 37 °C incubator with 5% CO2 atmosphere. Human PSCs were isolated from unidentified pancreatic cancer surgical specimens according to the protocol presented by Bachem et al.58. All experiments involving PSC were performed between passages 3 and 10. In brief, PSC was cultured in DMEM/F12 medium supplemented with 10% FBS and 1% penicillin/streptomycin in a 37 °C incubator with 5% CO2 atmosphere. All cell lines were regularly confirmed to be mycoplasma-free by PCR.
Preparation of conditioned medium
The medium was refreshed upon the cells met 50% confluence, and the conditioned medium (CM) was obtained by filtering the culture medium through 0.45 μm microporous membrane after 48 h of incubation8.
Western blotting
The cells were collected and lysed using RIPA buffer containing PMSF for 30 min on ice. The protein was obtained after centrifugation (12,000 g × 30 min) of the lysate at 4 °C, and its concentration was determined by a BCA protein assay kit (23227, Thermo, USA). Proteins (30 μg/each sample) were separated by electrophoresis on a 10% SDS-polyacrylamide gel at 120 V and transferred onto a PVDF membrane (Millipore, Germany) using a constant current of 250 mA for 2 h. The membranes were then blocked with 5% BSA for 1 h at room temperature solution before being co-incubated overnight at 4 °C with primary antibodies against β-actin (1:1000), CD71 (1:1000), GLUT1 (1:1000), p-p65 (1:1000) or p65 (1:1000). After being washed three times with TBS-T, the membranes were immediately incubated with a secondary antibody (goat anti-rabbit, 1:5000) at room temperature for 2 h. The visual blot images were acquired using ECL reagents on a Bio-Rad ChemiDoc Imaging System (Image Lab Touch Software version 2.4.0.03). Protein expression was normalized to β-actin or β-tubulin.
Confocal microscopy
For confocal microscopy assay, cells were washed twice with a washing buffer (D-PBS supplemented with 4.5 g/L of glucose and 5 mM of MgCl2), followed by treatment with Cy5-labeled AsiG (250 nM) either free or loaded by liposome with or without anti-CD71 antibody in a binding buffer (D-PBS supplemented with 4.5 g/L of glucose, 5 mM of MgCl2, 1 mg/mL BSA, and 0.1 mg/mL of yeast tRNA) for 1 h at pH 7.4 or 6.5 on ice. After two washes with the washing buffer, the samples were fixed in 4% paraformaldehyde for 10 min at room temperature. The cell membrane of T3M4 or BxPC-3 cells was stained using iFluor® 488-WGA dye, while the cell skeleton of PSC was stained using iFluor 488-labeled phalloidin. Prior to image visualization on a super-resolution confocal microscope (Leica SP8 STED 3X), the cell nuclei were counterstained with DAPI (1:1000) for 5 min.
Flow cytometry analysis
For flow cytometry assay, T3M4 or BxPC-3 cells were digested with 0.25% trypsin, counted, and divided into tubes containing 2 × 105 cells each. The cells were then incubated with different formulations containing 250 nM of Cy5-labeled AsiG in a binding buffer (200 μL) for 1 h on ice. Afterward, the unbound aptamer was washed out and cells were resuspended into 200 μL washing buffer to detect fluorescence intensity using a flow cytometry analyzer (Beckman, CytoFLEX S).
Cell viability
To determine the cell viability, T3M4 (6000/well), BxPC-3 (10,000/well) cells, and PSC were seeded in 96-well plates, respectively. They were then cocultured with indicated formulations at concentrations of 20 μM (PDAC cells) or 10 μM (PSC) of TPCA-1 and 50 nM of AsiG for a duration of 48 h at pH 7.4 or 6.5, followed by absorbance measurement at a wavelength of 450 nm with a microplate reader using Cell count kit-8 (CK04, DOJINDO, Japan). Relative cell viability was normalized to the control group at pH 7.4.
Immunofluorescence staining in vitro
T3M4 cells and PSC were seeded in gloss bottom dishes and received treatments of different formulations containing 20 μM (T3M4 cells) or 10 μM (PSC) of TPCA-1 and 50 nM of AsiG for 48 h at pH 6.5, followed by fixation in 4% paraformaldehyde for 10 min at room temperature. Prior to blocking the samples by 5% BSA for 30 min, permeabilization was achieved using 0.1% Triton X-100 for 15 min at room temperature. The primary antibodies against p65 (1:200), α-SMA (1:200), or fibronectin (1:200) were incubated overnight at 4 °C. Afterward, being washed with PBS, the cells were incubated with a secondary antibody (goat anti-rabbit, 1:1000) conjugated to either Alexa Fluor® 488 or Alexa Fluor® 594 for 1 h at room temperature. Following the counterstain of nuclei with DAPI (1:1000) for 5 min, images were visualized using a super-resolution confocal microscope (Leica SP8 STED 3X).
Sirius red staining
PSCs were seeded in a 6-well plate and subjected to different treatments for 48 h at pH 6.5. The deposited collagen was stained with 1 mg/mL Sirius red dye (S8060, Solarbio, China) dissolved in picric acid for 1 h. After being washed, the samples were imaged by a microscope.
RT-qPCR
T3M4 cells received formulation treatment at pH 6.5 for 48 h. Total RNA was extracted using RNAeasy™ animal RNA isolation kit (R0027, Beyotime, China) and reverse transcribed into cDNA using Hifair® III 1st strand cDNA synthesis super mix (11141ES60, YEASEN, China). RT-qPCR assay was implemented according to the protocol of the manufacturer using qPCR SYBR green master mix (11184ES08, YEASEN, China). The relative expression levels of Glut1 were normalized using β-actin. The primer sequences relative to RT-qPCR are listed in Supplementary Table 5.
Glucose uptake and Lactate secretion
For measurement of glucose uptake, Glucose Uptake Assay Kit-Red (UP03, DOJINDO, Japan) was utilized. Briefly, cells were seeded in 96-well plates and treated with standard culture medium or conditioned medium (50 μL of fresh standard culture medium and 150 μL of conditioned medium) at pH 6.5 for 48 h. After removing the supernatant, cells were washed with the pre-warmed glucose-free medium at 37 °C and subsequently incubated in a 37 °C incubator with 5% CO2 for 15 min. The pre-heated 2-DG probe solution was added and then incubated for an additional 15 min after removing the medium. Following washing with PBS, samples were analyzed using a fluorescent microplate reader (Ex: 540/15 nm, Em: 590/35 nm) with standardization of cell number by the Cell count normalization kit (C544, DOJINDO, Japan). To detect lactate concentration secreted by cells, the medium was collected to co-incubate with the working fluid of Lactate Assay Kit-WST (L256, DOJINDO, Japan) for 30 min according to the manufacturer’s instructions. The determination was carried out using a microplate reader at 450 nm. The cell number was also normalized. The lactate secretion was the difference in lactate concentrations in the culture medium before and after treatment with different formulations.
Extracellular acidification rate (ECAR) and oxygen consumption rate (OCR)
ECAR and OCR measurements were performed using the Seahorse XFe-96 analyzer (Agilent, USA). T3M4 cells (4000/well), BxPC-3 cells (7500/well), or PSC (2000/well) were plated overnight in the Seahorse XFe-96 Microplates and treated with different formulations in standard culture medium or conditioned medium at pH 6.5 for 48 h. Prior to the detection, the medium was replaced with Seahorse XF Base Medium (102353-100, Agilent, USA), supplemented with either glutamine (2 mM) for the glycolysis stress test or glutamine (2 mM), glucose (10 mM) and pyruvate concentration (1 mM) for the mitochondrial stress test, and subsequently incubated for an additional 30 min at 37 °C in a CO2-free incubator. For the ECAR measurements, the final concentration of glucose (10 mM), oligomycin (2 μM), and 2-DG (50 mM) of PDAC cells or glucose (10 mM), oligomycin (1 μM), and 2-DG (50 mM) of PSC were injected at indicated time points, respectively. For the OCR measurement, the final concentration of oligomycin (2 μM), FCCP (0.5 μM), antimycin A/ rotenone (1 μM/1 μM) of PDAC cells, and oligomycin (1 μM), FCCP (1 μM), antimycin A/ rotenone (1 μM/1 μM) of PSC were injected at indicated time points, respectively. At the experiment endpoint, the actual number of cells in the microplates was normalized by the Cell count normalization kit.
Establishing the orthotopic pancreatic cancer models
BALB/c nude mice (male, 8 weeks, 18-20 g) were acquired from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). A desmoplastic matrix orthotopic xenografts PDAC model was established by co-injecting 5 × 104 T3M4-luci cells and 1.5 × 105 PSC. Briefly, mice were anesthetized with isoflurane, and a small incision was made above the spleen to expose the pancreas. Cells were suspended in a 25 μL mixture of PBS and Matrigel (1:1, v/v) was injected into the pancreatic tails of mice. Then, absorbable sutures were used to close the wound. Growth of the orthotopic PDAC tumors was monitored by bioluminescence of luciferase using IVIS Lumina III In Vivo Imaging System. Mice were considered tumor-positive when the luminescence signal was higher than 105 p/s/cm2/sr. In accordance with relevant ethical guidelines, all animal protocols and experiments were approved by the Institutional Animal Care and Use Committee of the National Center for Nanoscience and Technology. Sex was not considered in this study because the dual rectification of metabolism abnormality of PDAC by the nanosystem is hardly influenced by sex. Male nude mice were used to construct an orthotopic pancreatic cancer model in current study. The nude mice were raised in pathogen-free conditions at a temperature of 20–22 oC and a humidity of 30–70%. Artificial light was set as 12 h of light and 12 h of darkness. Food and water were always available for the mice. The maximal tumor size permitted by the ethics committee was not exceed 10% baseline body weight (at the start of the study) in therapeutic studies, and none of the tumors in this study exceeded this size limit.
Circulation and biodistribution of T-AsiG-CPL in vivo
To investigate blood circulation and biodistribution of nanoparticles, siG-Cy5, AsiG-Cy5, and T-AsiG-Cy5-CPL (siG: 1.0 mg/kg) were intravenously administered to mice bearing orthotopic pancreatic tumors. Blood samples were collected at indicated time points post-injection. The circulation of the nanoparticle was determined by quantifying the fluorescence intensity of Cy5 in blood using IVIS Lumina III In Vivo Imaging System (PerkinElmer, USA). The same detection method was employed to investigate the biodistribution and accumulation of liposomes in tumors and major organs at 24 or 48 h post-administration.
Penetration of T-AsiG-CPL in tumors
To observe the penetration of nanoparticles in vivo, mice bearing orthotopic pancreatic tumors were randomly divided into two groups. One group received T-AsiG-CPL treatment 6 times (TPCA-1: 20 mg/kg, siG: 1.0 mg/kg, administered once every two days via intravenous injection), while the other group remained untreated as a control. At the end of therapy, each group was further randomized into two subgroups and given intravenous injections of siG-Cy5 or T-AsiG-Cy5-CPL, respectively. Mice were euthanized 24 or 48 h post-administration, and tumors were dissected to be immobilized on the device. Subsequently, Second Harmonic Generation (SHG) imaging was conducted at an excitation wavelength of 1150 nm using two-photon microscopy (Olympus, Japan). Sequential tomography consisting of 40 scans every three microns from the epidermis to the center of the tumor was performed. The three-dimensional reconstruction of the distribution and penetration of Cy5-labeled liposome deep within the tumor was further processed by FV315-SW software.
Treatment of tumor-bearing mice with T-AsiG-CPL in vivo
Growth of the orthotopic PDAC tumors was monitored by the bioluminescence of luciferase using IVIS Lumina III In Vivo Imaging System. Prior to each bioluminescence imaging session, D-Luciferin potassium salt (30 mg/mL) was intraperitoneally injected 10 min in advance. On the sixth day, nude mice were randomly divided into seven groups (n = 6 mice), as follows: Group 1: saline; Group 2: CPL; Group 3: TPCA-1; Group 4: T-CPL; Group 5: AsiG-CPL; Group 6: TPCA-1 + AsiG-CPL; Group 7: T-AsiG-CPL. All the reagents were administered intravenously at a unanimous dose of 20 mg/kg of TPCA-1 and 1.0 mg/kg of siG every other day. 48 h after the sixth administration, mice were euthanized by cervical dislocation, followed by the measurement of tumor weight and size (V = L × W2/2) at the experiment endpoint. In order to mitigate potential drug safety hazards, blood samples were collected for serum biochemical analysis, including typical liver function indicators (ALT and AST) as well as kidney function indicators (BUN and CREA). Following this, all tissues were fixed in 4% paraformaldehyde, paraffin-embedded, sectioned into 5 μm slices, and stained with Hematoxylin and Eosin (H&E). SHG images of collagen were visualized with H&E sections of tumors in a self-illuminating manner at 880 nm excitation wavelength using two-photon microscopy. Immunohistochemical staining for α-SMA (1:3000), fibronectin (1:200), p65 (1:200), and Ki67 (1:200) as well as IF staining for GLUT1 (1:200) was performed on tumor sections according to the standard protocols.
Statistical analysis
Statistical analysis was performed using GraphPad Prism (Version 7.00). The statistical significance was reported by one-way ANOVA or two-way ANOVA followed by Bonferroni’s multiple comparisons test, and the results were presented as mean ± standard error of the mean (SEM). p < 0.05 was defined as a significant difference. The number of samples (n) indicated biological replication, which is specified in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All remaining data can be found in the Article, Supplementary, and Source Data files. Source data are provided with this paper.
References
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Klein, A. P. Pancreatic cancer epidemiology: understanding the role of lifestyle and inherited risk factors. Nat. Rev. Gastroentrol. Hepat. 18, 493–502 (2021).
Encarnación-Rosado, J. & Kimmelman, A. C. Harnessing metabolic dependencies in pancreatic cancers. Nat. Rev. Gastroentrol. Hepat. 18, 482–492 (2021).
Halbrook, C. J. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell 31, 5–19 (2017).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009).
Kimmelman, A. C. & White, E. Autophagy and tumor metabolism. Cell Metab. 25, 1037–1043 (2017).
Parker, S. J. et al. Selective alanine transporter utilization creates a targetable metabolic niche in pancreatic cancer. Cancer Discov. 10, 1018–1037 (2020).
Sousa, C. M. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483 (2016).
Feig, C. et al. The pancreas cancer microenvironment. Clin. Cancer Res. 18, 4266–4276 (2012).
Provenzano, P. P. & Hingorani, S. R. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br. J. Cancer 108, 1–8 (2013).
DuFort, C. C., DelGiorno, K. E. & Hingorani, S. R. Mounting pressure in the microenvironment: fluids, solids, and cells in pancreatic ductal adenocarcinoma. Gastroenterology 150, 1545–1557.e2 (2016).
Koong, A. C. et al. Pancreatic tumors show high levels of hypoxia. Int. J. Radia. Oncol. 48, 919–922 (2000).
Kamphorst, J. J. et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 75, 544–553 (2015).
Lyssiotis, C. A. & Kimmelman, A. C. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 27, 863–875 (2017).
Pavlides, S. et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 8, 3984–4001 (2009).
Ligorio, M. et al. Stromal microenvironment shapes the intratumoral architecture of pancreatic cancer. Cell 178, 160–175.e27 (2019).
Capece, D. et al. NF-κB: blending metabolism, immunity, and inflammation. Trends Immunol. 43, 757–775 (2022).
Wang, X. et al. α-ketoglutarate-activated NF-κB signaling promotes compensatory glucose uptake and brain tumor development. Mol. Cell 76, 148–162.e7 (2019).
Capece, D. et al. NF-κB and mitochondria cross paths in cancer: mitochondrial metabolism and beyond. Semin. Cell Dev. Biol. 98, 118–128 (2020).
Zhao, W. et al. Galectin-3 mediates tumor cell–stroma interactions by activating pancreatic stellate cells to produce cytokines via integrin signaling. Gastroenterology 154, 1524–1537.e6 (2018).
Podolin, P. L. et al. Attenuation of murine collagen-induced arthritis by a novel, potent, selective small molecule inhibitor of IκB kinase 2, TPCA-1 (2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3- thiophenecarboxamide), occurs via reduction of proinflammatory cytokines and antigen-induced T cell proliferation. J. Pharmacol. Exp. Ther. 312, 373–381 (2005).
Hafez, I. M. & Cullis, P. R. Cholesteryl hemisuccinate exhibits pH sensitive polymorphic phase behavior. Biochim. Biophys. Acta 1463, 107–114 (2000).
Zhao, Y. et al. Tumor-specific pH-responsive peptide-modified pH-sensitive liposomes containing doxorubicin for enhancing glioma targeting and anti-tumor activity. J. Control. Release 222, 56–66 (2016).
Liu, G. et al. Engineering biomimetic platesomes for pH‐responsive drug delivery and enhanced antitumor activity. Adv. Mater. 31, 1900795 (2019).
Kawauchi, K., Araki, K., Tobiume, K. & Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-κB pathway and inhibits cell transformation. Nat. Cell Biol. 10, 611–618 (2008).
Özdemir, B. C. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734 (2014).
Baud, V. & Karin, M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 8, 33–40 (2009).
Di Conza, G., Tsai, C.-H. & Ho, P.-C. Fifty shades of α-ketoglutarate on cellular programming. Mol. Cell 76, 1–3 (2019).
Vonlaufen, A. et al. Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 68, 2085–2093 (2008).
Hwang, R. F. et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 68, 918–926 (2008).
Tsoi, K. M. et al. Mechanism of hard-nanomaterial clearance by the liver. Nat. Mater. 15, 1212–1221 (2016).
Ngo, W. et al. Why nanoparticles prefer liver macrophage cell uptake in vivo. Adv. Drug Deliv. Rev. 185, 114238 (2022).
Du, B. Transport and interactions of nanoparticles in the kidneys. Nat. Rev. Mater. 3, 358–374 (2018).
Connor, A. A. Pancreatic cancer evolution and heterogeneity: integrating omics and clinical data. Nat. Rev. Cancer 22, 131–142 (2022).
Neesse, A., Algül, H., Tuveson, D. A. & Gress, T. M. Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut 64, 1476–1484 (2015).
Schnittert, J., Bansal, R. & Prakash, J. Targeting pancreatic stellate cells in cancer. Trends Cancer 5, 128–142 (2019).
Hosein, A. N., Brekken, R. A. & Maitra, A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat. Rev. Gastroentrol. Hepat. 17, 487–505 (2020).
Masoud, R. et al. Targeting mitochondrial complex I overcomes chemoresistance in high OXPHOS pancreatic cancer. Cell Rep. Med. 1, 100143 (2020).
Wu, Q. et al. GLUT1 inhibition blocks growth of RB1-positive triple negative breast cancer. Nat. Commun. 11, 4205 (2020).
DeWaal, D. et al. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat. Commun. 9, 446 (2018).
Satriano, L. et al. Metabolic rearrangements in primary liver cancers: cause and consequences. Nat. Rev. Gastroentrol. Hepat. 16, 748–766 (2019).
Daemen, A. et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl Acad. Sci. USA 112, E4410–E4417 (2015).
Biancur, D. E. et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 8, 15965 (2017).
Qin, C. et al. Metabolism of pancreatic cancer: paving the way to better anticancer strategies. Mol. Cancer 19, 1–19 (2020).
Wang, Z. et al. Regulation of metabolism in pancreatic ductal adenocarcinoma via nanotechnology-enabled strategies. Cancer Lett. 560, 216138 (2023).
Xu, R. et al. Remodeling of mitochondrial metabolism by a mitochondria‐targeted RNAi nanoplatform for effective cancer therapy. Small 20, 2305923 (2023).
Li, X. et al. A “Valve‐Closing” starvation strategy for amplification of tumor‐specific chemotherapy. Adv. Sci. 10, 2104671 (2022).
Chen, F. et al. Extracellular vesicle-packaged HIF-1α-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat. Cell Biol. 21, 498–510 (2019).
Zhang, W. et al. Sustained release of TPCA‐1 from silk fibroin hydrogels preserves keratocyte phenotype and promotes corneal regeneration by inhibiting interleukin‐1 β signaling. Adv. Healthc. Mater. 9, 2000591 (2020).
Chen, Y. et al. IL-8 activates fibroblasts to promote the invasion of HNSCC cells via STAT3-MMP1. Cell Death Discov. 10, 65 (2024).
Han, X. et al. Reversal of pancreatic desmoplasia by re-educating stellate cells with a tumour microenvironment-activated nanosystem. Nat. Commun. 9, 3390 (2018).
Mauro, C. et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 13, 1272–1279 (2011).
Johnson, R. F., Witzel, I.-I. & Perkins, N. D. p53-dependent regulation of mitochondrial energy production by the RelA subunit of NF-κB. Cancer Res. 71, 5588–5597 (2011).
Helms, E., Onate, M. K. & Sherman, M. H. Fibroblast heterogeneity in the pancreatic tumor microenvironment. Cancer Discov. 10, 648–656 (2020).
Rebelo, R., Xavier, C. P. R., Giovannetti, E. & Vasconcelos, M. H. Fibroblasts in pancreatic cancer: molecular and clinical perspectives. Trends Mol. Med. 29, 439–453 (2023).
Boyd, L. N. C. et al. Heterogeneity and plasticity of cancer-associated fibroblasts in the pancreatic tumor microenvironment. Semin. Cancer Biol. 82, 184–196 (2022).
Pons, M., Foradada, M. & Estelrich, J. Liposomes obtained by the ethanol injection method. Int. J. Pharm. 95, 51–56 (1993).
Bachem, M. G. et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 115, 421–432 (1998).
Acknowledgements
This work was supported by the Beijing Natural Science Foundation (Z210017 to Y.L.), the National Key Research and Development Program of China (2021YFA1201103 to Y.L.).
Author information
Authors and Affiliations
Contributions
Y. Li, B. Wu, and G. Nie conceived the study and designed the experiments. B. Wu, Z. Wang, J. Liu, N. Li, X. Wang, H. Bai, and C. Wang performed the research. B. Wu, Z. Wang, and Y. Li collected and analyzed the data. G. Nie, J. Shi, S. Zhang, and J. Song, provided suggestions and technical support for the project. B. Wu and Y. Li wrote the manuscript. Y. Li and G. Nie supervised the project. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Shiv Singh, Bruce Shapiro, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, B., Wang, Z., Liu, J. et al. Dual rectification of metabolism abnormality in pancreatic cancer by a programmed nanomedicine. Nat Commun 15, 10526 (2024). https://doi.org/10.1038/s41467-024-54963-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54963-y
